cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;

地址:北京市丰台区西四环南路35号中都科技大厦10层
《互联网药品信息服务资格证书》 编号:(京)一非经营性一2021一0014
备案序号:京ICP备12047146号和京ICP备16030590号 信息产业部备案管理系统网址

扫
一
扫
© 2026北京远博康惠医药信息咨询有限公司 版权所有
北京远博医药CRO
感谢您关注远博医药微信公众号


© 2026北京远博恒康医药科技有限公司 版权所有
附件1
中药新药临床研究一般原则
一、概述
依据《药品注册管理办法》,为体现中医药特色,遵循中医药研究规律,继承传统,鼓励创新,进一步提高中药新药临床试验的水平和质量,推动中药新药的研究与发展,特制定本指导原则。
本指导原则是为中药新药临床试验的设计、实施和评价提供一般性方法学指导。
本指导原则是在2002年《中药新药临床研究指导原则(试行)》的基础上,基于中药新药临床试验现状,结合近年来中医药学、现代医学、临床流行病学、医学统计学等的新进展,参考国内外相关指导原则而制定。
本指导原则强调中药新药的临床试验需符合伦理学原则,充分保护受试者的安全;强调中药新药临床试验是以研究药物的临床价值为目标;强调在启动临床试验时,根据药物的潜在临床作用制定整体临床试验计划的重要性;强调临床试验过程应具有逻辑性,应重视早期探索性研究,不同阶段的各项临床试验应具有明确、具体的试验目的;强调临床试验过程中应重视获取的阶段性研究数据,不断地进行风险/受益评估,及时调整下一步研究计划,以降低研发风险。
本指导原则强调客观地评价中药新药的临床疗效及其特点,确证性临床试验有效性应以临床结局指标或公认的替代指标进行评价。强调重视中药新药的安全性研究,修订了心、肝、肾重要脏器安全性评价的具体要求,对于长期治疗不危及生命疾病的药物需延长疗程进行安全性研究。明确了中药新药开展风险/受益评估的要求、风险/受益评估的原则以及临床价值在风险/受益评估中的重要性。
本指导原则强调临床试验设计与实施过程中的质量控制,列出了需要关注的影响临床试验质量的常见因素,提出了中药新药临床试验用安慰剂研制的要求。
需要说明的是,本指导原则旨在提供一般性方法学指导,不排斥其他科学方法的合理应用。申请人如果能够以充分的证据说明本指导原则以外的方法具备科学性和合理性,同样可以获得认可。申请人还应关注疾病诊断、治疗方法的不断进步,药品注册相关法规的修订完善,在临床试验设计与实施过程中作出相应的调整。
二、伦理学及受试者的保护
尊重、保护受试者的权益、安全和健康是临床试验伦理学的基本原则。
中药新药临床试验的设计与实施应符合《赫尔辛基宣言》等国际公认的伦理原则,同时要符合我国药品注册相关法规、《药物临床试验质量管理规范》和《药物临床试验伦理审查工作指导原则》等相关伦理要求。
鉴于中医药的特点,中药新药临床试验伦理尚需要关注如下问题:药物组方与主治中医证候的方证相应问题;当中药与化学药物联合应用时,药物间相互作用所可能产生的安全性问题;有毒药材或长期临床使用的安全性问题等。
三、中药新药临床试验的特点
中药新药的研制是基于中医药理论和临床实践,中医证候的观察和疗效评价是中药新药临床试验的重要内容之一。中医证候既是目标适应症的纳入标准,同时也是疗效评价的指标。
中医证候的疗效评价方法应具有科学性,所获得的临床受益应具有公认的临床价值。
主治为病证结合的中药新药临床试验,主要疗效指标应选择临床结局指标或公认的替代指标。主要疗效指标如为改善症状、体征或疾病状态,提高患者生存质量,其临床价值应是公认的,并且应对疾病的临床转归无不利的影响。
中药新药临床试验需充分关注证候转化对药物有效性、安全性评价的影响。
中药复方制剂需注意方证相应,并针对预先拟定的中医证候进行评价。中药有效成份、有效部位制剂等需进行中医证候探索性研究,为Ⅲ期中医证候的确证性研究提供依据。
四、中药新药临床试验计划的制定及研发风险的控制
以注册为目的的新药临床试验应当是一个有逻辑、有步骤的过程,在这一过程中,早期小规模临床试验结果,为后续更大规模的、目的性更强的临床试验提供重要信息,用以进一步判断药物的临床价值和安全性风险。为了更好地降低药物研发风险,申请人在研发早期需根据药物的特点、立题依据及非临床研究的结果,拟定目标适应症的疾病发生发展演变规律,确定药物的临床定位、预期的临床价值和使用方法等,据此制定适宜的临床试验计划。申请人还应该意识到,药物无效或存在安全性问题,未必在临床前的研究阶段即显现出来。因此,即使进入临床试验阶段的药物能否成为上市的新药仍有待科学的临床研究与评价。
药物临床试验计划通常包括不同阶段及试验目的的临床试验方案,在每个阶段均需进行研发风险评估,包括临床定位是否准确、临床试验设计是否科学可行、临床试验质量控制是否良好等,并对计划适时作出调整,以规避药物研发风险。
(一)药物的临床定位
临床定位是指中药新药在拟定目标适应症中预期的治疗作用,该作用应具有公认的临床价值。客观、恰当的临床定位可以降低药物的研发风险。
确定药物的临床定位需考虑:适应症疾病发生发展演变规律;适应症疾病现阶段医学进展,所能达到的治疗水平,中医药目前在目标适应症治疗中的作用和地位及药物潜在的临床价值;需明确是治疗用药还是预防用药,是影响疾病进程还是改善症状,是联合现有治疗方法还是单独使用等。
(二)临床试验设计方法的科学性
在中药新药确证性临床试验设计中,主要疗效指标必须明确,如果主要疗效指标不明确或者试验结束后才确定或更换主要疗效指标都会带来有效性评价的问题。主要疗效指标如采用替代指标,应预先设定,且应被广泛认可。
在进行等效性或非劣效性试验时,非劣效或等效“界值”的确定应有充分的依据,如果依据不足,将可能无法证明药物的有效性。
对临床需要长期或反复使用的药物,应有足够的样本量及药物的暴露时间和暴露剂量,以充分观察药物的安全性。
(三)早期探索性临床试验
加强早期探索性临床试验的研究,将有助于及时评估临床定位是否恰当、临床受益大小、给药剂量和疗程的合理性、预期或非预期不良反应严重程度和发生率等方面是否存在问题,以降低后期研发风险。
(四)临床试验质量控制
临床试验设计与实施过程应考虑到临床试验质量控制不佳所带来的研发风险。诸如临床试验预先未设计导入期,对关键实验室检查指标未预先建立一致性检测要求;未按随机要求入组,未按规定使用合并用药,研究者对疗效观测指标测量方法的培训欠缺等,均将影响临床试验质量,导致临床试验结果不能支持其有效性、安全性评价。
五、风险/受益评估
风险/受益评估,一般是指受试者使用受试药物以后所能获得的治疗方面的受益与所承担的风险之间平衡的把握,应该充分考虑临床价值在风险/受益评估中的重要性,以评价其是否具有上市价值。
受益主要体现在所治疗疾病的病情程度的改善,疾病持续时间的缩短,生命维持时间的延长和生存质量的提高等方面。风险主要包括不良反应的类型、严重程度、持续时间及发生频率等。
(一)开展风险/受益评估的要求
1.完整的数据库
临床试验的安全性数据库越大、越全面,发现严重不良事件/不良反应的可能性就越大。风险/受益评估至少应具备一个科学的、大小合理的、完整的、可供评价的安全性数据库。安全性数据库的合理大小取决于诸多因素,如中药新药的创新程度、现有
治疗药物的情况及与其相比的安全性,治疗的目标适应症人群受益程度、疗程长短等。
2.合理的对照
合理的对照能够获取比较性的疗效和安全性数据,有助于分析药物疗效和不良反应是药物所致还是由于其他因素所致。
采用安慰剂对照不仅可以了解药物的“绝对”疗效,便于清晰地评价安全性(区分药物本身所致不良反应以及来自潜在疾病或并发疾病的不良反应等),还可以检测试验本身的灵敏度。
采用阳性药物对照有助于获得伦理学的批准,可获得中药新药与已上市公认有效药物的“相对”有效性和安全性。
采用安慰剂和阳性药物对照的三臂试验设计具备许多优点,不仅可以获得药物的“绝对”疗效以及试验检测灵敏度的内部证据,还可以同时进行与阳性药物疗效等方面的对比研究。
在符合伦理学的前提下,应有安慰剂对照的临床试验数据,以进行风险/受益评估。采用三臂试验设计的临床试验数据,则有助于判断临床价值,以获得更好的支持上市的证据。
3.足够的随访
应对所有的受试者访视到试验结束,必要时甚至随访到试验结束之后。特别是在需要长期治疗的研究中,足够长时间的随访对确定其安全性非常关键。随访应包括脱落病例及因达到了观测
的主要结局而提前完成研究的受试者。随访的持续时间需要根据药物的具体情况而定。
4.查实退出研究的原因
不论是研究者决定的退出还是受试者决定的退出,都应查明退出的原因,尤其关注是否因为存在安全性问题。应对因安全性问题而退出者持续随访到不良事件完全消除或稳定(医学上认为可以停止观察)为止,并在研究病历和病例报告表中记录随访数据。对研究过程中受试者出现异常的实验室数值,即使尚未达到规定的不良事件的程度,也应当全面收集相关信息,并加以记录和必要的随访。应当要求退出研究的受试者继续提供有关信息,以判断是否为严重或显著安全性问题,否则将影响对药物安全性的科学评价。
(二)风险/受益评估重点
针对所完成临床试验的有效性、安全性数据,需整体评价是否具备风险/受益评估的条件,在此基础上方可进行风险/受益评估。临床试验总结报告中应提供风险/受益评估的分析与结论。
1.风险评估重点
对各期临床试验过程中出现的全部的不良事件和严重不良事件等进行合理的因果判断,以不良反应类型、发生率和严重程度等来评价药物的安全性风险。还需要关注安全性数据是否完整充分、有无遗漏;发生的风险是否与对照组进行合理的比较,是否包括少见的、非预期的、严重的及剂量相关的不良反应,有无存在同类药物的安全性问题等。
2.受益评估重点
对各期临床试验的全部有效性结果进行合理分析,各期临床试验有效性结果应为具有逻辑的证据链。应以公认的临床结局指标或替代指标的结果来评价药物带来的受益。评价内容包括试验的纳入人群是否代表方案中的目标适应症人群,是否有合理的对照,主要疗效指标是否恰当,统计分析方法是否正确,研究结论是否确切,主要疗效指标与次要疗效指标的有效性结果之间的一致性等。
(三)风险/受益评估原则
针对不同的疾病、同种疾病不同的严重程度,对于治疗所期望的疗效以及可以承受的风险是不同的。
应该区分药物针对疾病某一方面的有效性和患者的受益这两个概念,两者既有关联又有区别。患者的受益需要以有效性作为基础,但对疾病某一方面有效的药物未必使该疾病患者受益。受益评估更看重的是临床结局指标或公认的替代指标的结果所体现的临床价值。
对于安全性的可接受性,会因受益大小、疾病类型和严重程度的不同而变化,其风险/受益的平衡点是不同的。
在药物的风险/受益评估中,通常不能仅仅局限于所研发药物的安全性和有效性,还应该兼顾与现有的治疗药物、治疗水平进行必要的比较,依据其受益所体现的临床价值的大小,其风险是否可以接受,以综合评估是否受益大于风险,具备上市价值。
相对于疾病对人体的危害及现有治疗方法及水平,药物的疗效与对照药物相比体现出优势或特色并能够使患者受益且具有临床价值,如果药物的不良反应可接受,则具有上市价值;如果药物的不良反应比较大,则需要结合适应症特点、适应人群的特点、疾病本身对人体的危害及其现有治疗药物和治疗方法、不良反应是否可控、可防等因素,综合评估其是否具有上市价值。
药物的疗效与对照药物相比未体现出优势或特色,或虽体现出优势或特色,但相对于现有治疗方法及水平,患者受益的临床价值不大,如果药物的不良反应比较大,则不具有上市价值;如果药物的不良反应可接受,申请人应充分评估其是否具有上市价值,否则应考虑继续研发的必要性。
药物风险/受益评估是一个动态的过程。申请人应该通过不断的研究,在不同阶段的临床试验结束时都进行风险/受益的评估,以及时评估药物风险的性质和与受益关联的风险程度。在受益的基础上通过风险控制计划,尽量减少风险,努力达到受益最大化。
药物获准上市后,还需通过Ⅳ期临床试验等进行上市后再评价,以考察在广泛使用条件下的药物的疗效和不良反应,尤其发现罕见的、非预期的、特殊的、严重的、长期使用时出现的不良反应。当收集到更多的有效性、安全性数据时,需要对风险/受益重新作出评估,如果风险大于受益,即使药物已经上市也应撤市。
六、中药新药Ⅰ期人体耐受性试验设计
I期临床试验是对药物进行初步的临床药理学及人体安全性评价试验,包括人体耐受性试验和人体药代动力学等研究。
人体耐受性试验是观察人体对于药物的耐受程度,其目的是为Ⅱ期临床试验确定合适的剂量,为用药间隔和疗程等提供依据。中药新药的人体药代动力学研究应参照化学药品关于药代动力学的技术要求,本指导原则中不再涉及相关内容。
(一)I期人体耐受性试验设计要点
I期人体耐受性试验首先要进行单次给药耐受性试验,针对药物的用药剂量进行探索研究,在此基础上确定是否进行连续给药耐受性试验。
1.单次给药耐受性试验
(1)受试者
一般选择健康志愿者,年龄:18岁~50岁,同批受试者年龄相差不宜超过10岁。男女数量最好相等。女性受试者应排除
月经期、妊娠期、哺乳期。体格检查包括体重、身高符合标准范围,无阳性体征发现,如心、肺听诊及血压正常,胸、腹部叩、触诊无异常发现,无明显的皮下淋巴结肿大等。实验室检查项目包括:血、尿、大便常规。除本指导原则要求的必须观察的实验室检查项目外,还应进行尽量全面的血液生化学检查、凝血相关指标、传染性疾病筛查以及胸片、B超、心电图(ECG)等检查。上述检查结果均应在正常范围内。
妇产科药物的Ⅰ期临床试验应选择月经规律的近期无生育需求的女性作为受试者。除非是儿科方面的特殊需要,儿童不作为受试者。毒性较大或耐受性在正常人与患者间差异较大的药物,可以选择心、肝、肾、血液功能基本正常的轻型患者为受试者。
制定排除标准时应考虑:近期参加过其他临床试验;近期曾应用各种药物者(包括中药);3个月内用过已知对某脏器有损害的药物或目前正在使用药物者;3个月内有献血史;有药物过敏史或过敏性体质者;有心、肝、肾等病史者;与受试药物作用有关病史者;有其他影响药物吸收、分布、代谢和排泄等因素者。
(2)试验设计
I期临床试验可以是开放、基线对照的。为避免干扰,鼓励采用随机化和盲法等设计,以排除受试者之间的主观症状的
相互影响和研究者判断症状时的主观因素影响,以及实验室检查指标波动的影响,以提高观察结果的可靠性。
有时为了判明临床试验中出现的某些不良反应确是由于药物所引起,而不是受试者的心理作用或其他非药物性因素(如环境或生理性波动等)所致,可设置安慰剂对照以助于说明问题。
(3)试验例数
I期临床试验单次给药耐受性试验例数的确定需根据药物的特点、药理毒理研究结果所提示预计的安全性范围、预期活性、拟定的目标适应症情况等确定。从初试起始剂量到最大变量之间设若干组。各个试验组剂量由小到大逐组进行,一般先由低剂量开始,每剂量2~3人,接近治疗量后,每组6~8人。在低剂量组,病例数可以适当减少,随着剂量的增加,则受试者数量逐渐递增,递增的目的是便于尽快发现不良反应。如果药物的活性较强或毒性较大时,剂量递增梯度应缩小,可多设几个组,并增加试验例数。
(4)给药途径
根据药物的立题依据确定给药途径。口服给药者,一般应在禁食12小时后空腹服药。
(5)观察指标
观察指标要全面,除必须进行的临床症状、生命体征观察及实验室检查外,还应该根据药物既往人用经验所提示的毒性、非临床安全性研究所明确或提示的毒性靶器官、同类药物的毒性靶器官等增加一些特殊观察指标,以及增加临床前所提示的预期的药理作用的指标。
①常规检查指标:临床症状,体征,实验室检查(范围包括心血管、呼吸、消化、泌尿、内分泌、血液系统等)。外用制剂局部用药物应注重观察局部刺激症状,注射剂还应重点观察过敏反应等。用药前后均应作详细记录。
②特殊检查指标:指为观察药物可能存在的不良反应而做的检查。应针对非临床安全性研究结果设计相应的安全性观察指标。
③预期药理作用指标:如心血管药物,应详细观察对血压等的影响;妇科调经药物,需观察对妇女月经周期相关指标的影响等。
(6)观察时点
应根据具体药物特点和给药方法的不同来确定。在试验前1周内或尽可能在短时间内完成全部检查指标,给药后应密切观察受试者的一般情况、呼吸、心率、血压、体温及ECG等,观察时点应根据具体药物特点来确定。
一般给药后24小时、72小时观察全部指标,个别药物可观察2~5天。出现不良反应者应追踪随访,直到恢复正常。
注射剂从给药开始即需密切观察受试者可能出现的不良反应,并增加相关安全性指标的检测时点。
(7)试验终止指标
耐受性试验不仅要找出不出现不良反应的剂量,还应了解出现轻度不良反应的剂量及其性质。根据适应症的不同,应预先规定出现何种程度的不良反应时作为试验终止指标。通常以受试者出现半数轻度不良反应为试验终止指标,对于抗肿瘤药物等可规定出现较严重的毒性反应作为试验终止指标。
在剂量递增过程中如出现了不良反应,虽未达到设计的最大剂量,亦应终止试验。在达到最大剂量仍无不良反应时,试验即可结束。
(8)剂量设计
根据药理毒理研究结果首先设计“起始剂量”及“最大剂量”。剂量确定应当慎重,以保证受试者安全为原则。
起始剂量设计,下述四种方法可供参考。
①Blach well法:最敏感动物药物单次给药毒性的LD50的1/600或最小有效剂量的1/60以下。
②改良Blach well法(考虑安全性):两种动物药物单次给药毒性LD50的1/600及两种动物药物重复给药毒性的有毒剂量的1/60以下,本法考虑了非临床研究4种试验(包括药物单次给药毒性和药物重复给药毒性)的安全因素,较为妥善,是目前常用的方案。
③Dollry法(考虑有效性):最敏感动物最小有效剂量的1/50~1/100或同类药物临床治疗剂量的1/10以下。
④改良Fibonacci法(起始剂量较大,用于抗肿瘤药物):小鼠药物单次给药毒性LD10的1/100或大动物最低毒性剂量的1/40~1/30为起始剂量。本方法简单易行,曾较多用。但只凭一二种动物进行估算,LD10及最低毒性剂量变动幅度较大。
由于药物的不同,选择起始剂量的方法也不一样,没有固定模式,应视具体情况而定。对那些有明显药理活性的中药新药,起始剂量还应更小。中药注射剂过敏反应有时在极低剂量即可出现,切不可机械地按动物的剂量折算为人用剂量。
最大剂量的估算,可参考临床应用该类药物单次最大剂量设定。下述二种方法亦可供参考:①动物在药物重复给药毒性研究中引起中毒症状或脏器出现可逆性变化的剂量的1/10。②动物在药物重复给药毒性研究中最大耐受量的1/5~1/2。
如试验达到最大剂量受试者仍无不良反应时,试验即可结束。剂量递增到出现终止指标或其他较严重的不良反应时,虽未达到最大剂量,也应终止试验。
(9)剂量梯度
在“起始剂量”及“最大剂量”的范围内,按递增比例分若干个剂量级别,剂量级别的多少需视药物的安全范围大小,根据需要而定,一般不少于5个剂量组。先由低剂量开始,每剂量2~3人;接近治疗量后,每组6~8人;在达到最大剂量仍无不良反应时,一般即可终止试验,并以此为最大耐受量。耐受性试验时,每名受试者只能接受一个剂量的试验,不得对同一受试者进行剂量递增与累积耐受性试验,以确保受试者安全。每个剂量需要一组受试者,要在一个剂量组试验结束后才能进行下一个剂量组的试验。
通常低剂量试验如果安全性可靠,再进行下一个剂量的试验,依此类推。高风险品种(如中药注射剂),建议对起始剂量组受试者逐例进行试验。
2.连续给药耐受性试验
连续给药耐受性试验是在单次给药的基础上,根据药物临床使用的疗程,以剂量递增的方式分不同的组别连续给药,以对药物进行更进一步的耐受性和安全性评价。
连续给药的研究方法与单次给药基本相同,但需注意如下问题:
(1)受试者的选择:由于连续给药的疗程较单次给药长,因此药物本身对受试者健康的影响更复杂,如果药物对生育、免疫功能等方面有影响的话,可考虑以相关疾病轻型患者为主。
(2)剂量设计:连续给药耐受性试验通常至少应进行2个剂量组,每组6~8人。给药剂量为单次给药耐受性试验未出现不良反应的最大剂量(称为“最大耐受量”)下降1个剂量进行连续给药耐受性试验。如试验中出现明显的不良反应,则再下降1个剂量进行另一组试验;如试验中未见明显的不良反应,即上升一个剂量(即用最大耐受量)进行耐受性试验。
(3)疗程:当药物所拟定适应症预计临床治疗需要长期给药时(如连续治疗6个月或以上,或者间断治疗的累计时间大于6个月),除非受药物的毒性或药理作用所禁忌,连续给药耐受性试验建议不少于4周。
(二)不良事件/不良反应的观察与判断
必须确保受试者的安全,在试验期间必须对所有不良事件进行监测并详细加以记录,同时为发生任何不良事件/不良反应的受试者提供有效的医疗处理。对不良事件的判断不仅要回答是否与药物有关,还要考虑是否与剂量相关。
鉴于中药有性味、归经的特殊性,Ⅰ期临床试验也可研究与中药药性相关的不良反应。
(三)Ⅰ期耐受性试验的总结要点
由负责Ⅰ期耐受性试验的主要研究者作出总结。总结内容主要包括:未发生不良反应的剂量;发生不良反应的剂量;不良反应的表现,发生时间,持续时间,有无前期征兆等。如发现个别受试者出现的严重不良事件/重要不良事件确属药物所致,应及时进行剂量相关性分析。对不良反应的转归应注意观察是渐次加重还是自行缓解。对不良反应分析应详细列表。
需明确I期临床试验的安全剂量,结合人体药代动力学试验结果(如有),推荐Ⅱ期临床试验的剂量、给药方案、安全性指标等。在推荐Ⅱ期临床试验的剂量时也应考虑不良反应的危害程度。不良反应危害较轻、易耐受、不需处理、不影响日常活动者,可推荐较大剂量。不良反应的危害性大的药物,尽管用药剂量在耐受量以下,考虑到个体差异,一旦发生,后果严重,则应推荐较小剂量。
(四)Ⅰ期耐受性试验中应注意的问题
1.如I期耐受性试验的“最大剂量”设计过低,未达到药物实际的最大耐受量,将导致得出的Ⅱ期临床试验推荐剂量偏低,可能直接影响Ⅱ期临床试验药物的有效性。
2.耐受性试验由于受试者例数较少,因此对受试者在受试过程中出现的变化,逐例进行专业分析显得十分重要。有些检测数据变化虽在正常参考值范围内,也应从专业上仔细判断是否有临床意义。
3.应该充分认识到实验动物与人之间的种属差异,以及毒理研究所提供的安全剂量与人体试验存在差距的可能性,对安全性指标的设计要全面并科学评价。
4.由于某些受试者受到试验环境及知情同意书等的暗示影响,可能会出现头昏、乏力或胃肠反应等症状,如果采用随机、
盲法、安慰剂对照等设计,会有利于排除上述干扰,对耐受性试验结果作出正确判断。
5.I期耐受性试验如果是以患者为受试者,在不增加受试者痛苦的前提下,可同时进行一些无创伤性检查以观察药效。
七、中药新药临床试验设计的一般考虑
如果I期临床试验结果支持后续的临床试验,则应根据临床试验计划,分阶段通过多个不同目的的临床试验逐步探索和确证药物的有效性和安全性。本章节重点阐述设计一项临床试验方案时应考虑的主要内容,包括临床试验目的、目标适应症人群、试验设计方法,有效性和安全性评价指标等。
确定一项中药新药的临床试验目的,需依据临床试验计划整体考虑,明确该项临床试验是探索性试验或确证性试验,应充分评估既往药理毒理研究或临床试验所提供的数据,明确拟开展临床试验需回答的问题,并注意各项临床试验之间合理有序地衔接。
试验目的是设计和制定临床试验方案的前提。
1.确立试验目的的原则
一项临床试验设计一般确定一个主要目的,根据需要有时可以有次要试验目的。需要根据试验目的明确主要疗效指标、次要疗效指标及安全性指标等。
2.确定试验目的的依据
(1)处方组成与拟定的功能主治,既往临床应用及研究基础。
(2)药理、毒理研究结果。
(3)前期的临床试验结果。
(4)药品监督管理部门的相关要求。
3.试验目的的表述
试验目的的表述应清晰明确,主要围绕受试者、设计方法、探索性临床试验/确证性临床试验,有效性和安全性指标等表述,注意突出主要试验目的。如针对一个中药复方制剂,适应症拟定为2型糖尿病(气阴两虚证),试验目的表述为“以糖化血红蛋白复常率为主要疗效指标,采用随机、双盲、安慰剂对照、多中心临床试验设计,评价受试药物用于治疗2型糖尿病(气阴两虚证)有效性和安全性的确证性临床试验”。
1.临床试验设计的基本原则
临床试验设计时必须遵循对照、随机和重复的原则,这些原则是减少临床试验偏倚的基本保障。
(1)对照
为了评价一个药物的疗效和安全性,必须设立可供比较的对照。常用的对照有安慰剂对照、阳性药对照、剂量对照等。
(2)随机
随机是指参加临床试验的每一个受试者都有相同机会进入试验组和对照组。随机化有利于避免选择性偏倚,使得受试者进入试验组或对照组是随机的,从而保证各种影响疗效评价和安全性评价的因素(已知或未知)在不同组别中分布均衡,保证了不同组别间的受试者的可比性。
(3)重复
重复是指在相同试验条件下独立重复试验的次数,在临床试验中指各组受试者的数量。足够多的重复可以增加试验的可靠性,从而正确地反映药物的疗效和安全性。样本量的计算方法可参照本原则中“样本量”的有关要求和相关生物统计学指导原则。
2.临床试验设计的基本方法
(1)随机化
随机化方法通常分为三类:完全随机化、限制性随机化和适应性随机化。通常用计算机编程来产生随机分组方案。随机分组方案需有重现性。
①完全随机化:除了对受试者数量以及各试验组之间受试者的分配比例有限制外,对随机化序列的产生不加任何限制。
②限制性随机化:主要包括分层、区组随机,是临床试验中最常用的方法。分层因素应根据试验目的和影响试验结果的因素来确定,如试验中心、疾病亚型等都可作为分层因素考虑。分层对于组间均衡性是有帮助的,但受试者数过少时,层数不宜过多,否则将给试验实施和统计带来困难。区组(即分段)随机是按区组随机地纳入受试者,同一区组内的受试者由于接受治疗的时间相近,当药物的疗效与季节或时间趋势有关时,有助于增加各组的可比性。当样本大小、分层因素及区组长度确定后,由生物统计学专业人员在计算机上使用统计软件产生随机数字表,并据此得到分组方案。
③协变量适应性随机化:也称为动态随机化,是依据影响临床治疗效果的预后因子(协变量)当前在各组的分布情况,调整分组概率,以控制协变量在各组的平衡。
无论应用何种随机化方法,均应重视随机隐藏,没有随机隐藏的随机实施过程不是真正的随机化。
(2)盲法
盲法是为了控制试验过程中的各种偏倚,包括评价偏倚、统计分析时的解释偏倚等。临床试验根据设盲的程度分为开放(非盲)、单盲、双盲。双盲试验需要试验中所采用的处理方法在用药前或用药时都无法从感官上识别出来,且在整个试验过程中都保持盲态。
如果基于伦理学和可行性的考虑,不适宜采用双盲,则应考虑单盲试验或开放试验。此类试验需要注意避免由于临床试验参与人员可能知道受试者的随机化分组情况,而影响进入试验的受试者分组。同时,在此类试验中由于受试者知晓所接受的治疗,他们可能从心理上对治疗作出相应的反应,对试验结果产生偏倚。即使是疗效观测指标属于客观的指标,如生存率、病死率等,对于研究者而言,如果知晓受试者的治疗措施,则对于受试者死因的确定,以及死因的诊断等都有可能引起偏倚。所以,采用单盲或开放试验均应制订相应的控制偏倚的措施,使已知的偏倚达到最小。
另外,当受试药物和对照药物的剂型、用法用量不同时,则采用模拟技术,如双盲双模拟技术,即为受试药物与对照药物各准备一种安慰剂,以达到试验组与对照组在用药的外观与给药方法上的一致。
应在试验方案中说明采用不同设盲方法的理由,以及通过其他方法使偏倚达到最小的措施。
盲法试验需要保留盲法操作过程文件的记录,并在临床试验总结报告中说明,以附件作为药品注册申请文件提交。
(3)多中心临床试验
多中心临床试验是指由一个主要研究者总负责,多个临床试验机构合作,按同一临床试验方案同时进行的临床试验。多中心试验可以在较短时间内招募试验所需的受试者,且受试者范围广,用药的临床条件广泛,试验的结果对将来的应用更具代表性。
多中心临床试验要求不同中心的研究者采用相同的试验方法,所以试验过程要有严格的质量控制。
3.临床试验设计的基本类型
在临床试验设计方案中,统计设计类型的选择是至关重要的,因为它决定了样本量的估计、研究过程及其质量控制。因此,应根据试验目的和试验条件的不同,选择不同统计设计方法。
(1)平行组设计
平行组设计是指将受试者随机地分配到试验的各组,同时进行临床试验。平行对照不一定只有试验组和对照组两个组别,可为受试药物设置多个对照组,受试药物也可按若干剂量分组。对照组的选择应符合设计方案的要求。本设计的优点是有利于贯彻随机化的原则,避免非处理因素的影响,增强试验组和对照组的可比性,控制试验误差和偏性。
(2)交叉设计
交叉设计是一种特殊的自身对照设计,将每个受试者随机地在两个或多个不同试验阶段接受指定的处理(受试药物和对照药物)。这种设计有利于控制个体间的差异,减少受试者人数。最简单的交叉设计是2×2形式(AB/BA)。每个受试者需经历如下几个试验过程,即筛选期、第一试验阶段、洗脱期、第二试验阶段。在两个试验阶段分别观察两种药物的疗效和安全性。交叉设计资料分析时易于混杂延滞效应(前一个试验阶段处理效应对后一阶段试验的影响)。由于2×2交叉设计不能检测延滞效应,使用该设计需说明如何消除延滞效应,或可采用重复交叉设计,例如ABBA/BAAB设计。每个试验阶段后需安排足够长的洗脱期(5至7个药物消除半衰期),以消除前一阶段的延滞效应对后一阶段试验的影响。交叉设计要求每个阶段的病情经恰当的洗脱后具有可比性,多用于控制病情的药物的临床试验,对于进行性疾病或有望治愈的疾病不能使用交叉设计。
(3)析因设计
析因设计是将试验中涉及的各因素的所有水平进行完全交叉而形成份组的试验设计,用于检验各因素间是否存在交互作用,或通过比较找出最佳组合,或比较各因素不同水平的效应大小。析因设计缺点在于当因素过多或因素的水平数过多时,分组较多,因此所需要的样本量太多。所以,进行析因设计一般要求处理因素最好在4个以内, 各因素包括的水平数也不宜划分得过多。
(4)成组序贯设计
成组序贯设计是将整个临床试验分成几批,逐批序贯进行,每一批受试者试验结束后,及时对主要变量(包括有效性和安全性)进行分析,一旦可以得出结论(无效结论或有效结论)即停止试验。每一批受试者中试验组与对照组的例数相等或比例相同,且不宜太少,批次以不大于5为宜,以减少多次揭盲带来的α消耗。统计学方法必须事先说明关于处理结果和病例所指定的处理(如破盲)信息的可获得性。该设计的优势在于当受试药物
的疗效明显优于对照药物或安全性风险明显高于对照药物时,可以较早终止临床试验,缩短试验时间,减少受试者的数量和风险暴露的时间。受试者以序贯的方式分批入组,对临床观察结果定期进行评估,盲底要求一次产生,分批揭盲和分析。成组序贯设计是一种方便的进行期中分析的方法。尽管成组序贯设计不是唯一的可用于期中分析的方法,但它应用得最广泛。成组序贯的实施要求由申请人设立一个独立的数据安全监查委员会,定期对研究进展、安全性数据和有效性终点进行评估,向申请人建议是否继续或停止试验。
(5)加载设计
加载设计是联合治疗设计的一种方法,当所研究的疾病已经有一种标准治疗并且被证实能够降低该病的病死率、复发率等时,基于伦理学原则,临床试验时一般不宜中断原来的标准治疗,只能继续保持。如果使用安慰剂的盲法对照设计,则所有受试者在接受这种标准治疗的基础上,随机给予受试药物和安慰剂治疗。
由于加载设计通常是在现有临床标准治疗基础上加上受试药物或安慰剂,得到的疗效是多种施加因素的结果,必然给受试药物的疗效确认带来困难。一般在临床试验中仅采用安慰剂对照难以实施,或仅以标准治疗作阳性对照难以评价,为了保护受试者,客观评价药物的真实效应时可考虑加载设计。如果联合用药方能体现中药新药的临床价值,也可以采用加载试验设计。
在采用加载设计时,所选择的标准治疗应被公认,疗效指标要明确和恰当,应能反映出所加载药物的作用。受试者选择应有可比性,一般筛选出既往使用标准治疗已经取得最大疗效,但未达到治疗目标,同时病情保持稳定的目标适应症人群作为受试者。
在采用加载设计时,应注意标准治疗的标准化和一致性,其中包括规定允许标准治疗的条件,允许使用药物的种类及其剂量、方法,使用的时间等。观测指标选择应全面,除了评价受试者疾病主要疗效指标外,有时一些标准治疗药物的耗用量或使用频率,某些标准治疗已知不良反应发生的频率或严重程度的改变,也可能作为评价药物作用的指标。
当标准治疗所用药物的作用机制与受试药物不同时,加载设计研究显得更加有效。由于加载设计取得的是一种联合治疗效果,对受试药物作用的评价要恰如其分。同时需要注意的是,加载试验设计还需证明没有影响或干扰标准治疗的有效性。
加载设计的缺陷:①由于是多种药物同时使用,容易受到混杂偏倚的影响。②出现罕见或不常见的不良反应时,往往无法确定是由哪种药物或两种药物共同造成的,受试者需要承担两种药物未知的混合作用的风险,解释有时显得较为复杂或困难。③如标准治疗本身的疗效过高,由于“天花板”效应导致无法鉴别药物的疗效。
加载设计由于存在以上缺陷,故使用时需慎重。
(6)剂量-效应研究设计
中药有效成份和有效部位制剂等需进行剂量-效应关系研究。中药复方制剂一般也应进行剂量-效应关系研究。
中药新药剂量-效应的探索性临床试验通常在Ⅱ期临床试验中完成,其研究设计的类型一般有平行量效研究、交叉量效研究、强制剂量滴定和供选择的剂量滴定等。
平行量效研究是剂量研究中的常用设计方法。即随机平行的剂量—效应研究,把受试者随机分为数个有各自固定剂量的组。固定剂量指最终的或维持的剂量;受试者可开始时即用此剂量,也可以安全地逐渐滴定到此剂量(通常是通过强制的滴定方案)。在以上二种情况下,最终剂量应维持足够的时间来进行量效关系比较研究。
在平行量效研究中,中药有效成份、中药有效部位的制剂应设置多个剂量组,通过试验获得剂量-效应曲线,以证明剂量-效应关系;中药复方制剂除安慰剂组以外至少应有2~3个剂量组。
在平行量效研究中,即使未设立对照,也可以进行剂量-效应研究。值得注意的是,如果选择的多个剂量过大或剂量组间剂量梯度过小,则有可能导致不能形成量效曲线,无法获得量效关系。在此情况下,如果试验中设置了安慰剂对照,并且某剂量组与安慰剂组效应差别有统计学意义,则可以说明药物存在量效关系。因此,建议在符合伦理的前提下使用安慰剂对照。另外,增加阳性对照也可以为剂量的确定提供一定的依据。
一般情况下,剂量-效应关系临床试验要求各剂量组的效应形成较完整的量效曲线,量效曲线一般采用曲线拟合的方法获得,拟合的曲线应有统计学意义。一般不要求各剂量组间两两比较显示出统计学差异。对于中药复方制剂,由于剂量组设置相对较少,则应采用组间效应两两比较来确定量效关系,最终推荐的最佳剂量其效应与其他剂量组的效应比较,至少有一个剂量组的差异应该表现出有统计学意义的趋势。
一般而言,设置的剂量组越多、剂量梯度越合理,每组所需的样本量越小,反之所需的样本量则越多。
此外也可以选择交叉量效研究、强制剂量滴定等试验设计方法进行剂量-效应研究。
选择合格受试者,是设计和实施临床试验的重要环节。受试者的选择是根据临床试验目的来决定的,恰当的疾病与中医证候诊断标准是确保样本同质的关键。尤其是多中心临床试验,为了选择合适的受试者,试验设计中应确定统一的目标适应症受试者诊断标准(包括疾病与中医证候)、入选标准、排除标准、退出试验标准、剔除病例标准等。
1.受试者选择标准
(1)诊断标准
临床试验设计时应根据所确定的适应症,分别列出西医、中医诊断标准及中医证候辨证标准,并注明诊断标准的来源,如国际、国内标准,包括政府主管部门、全国性学术组织制订的诊断标准和权威性著作标准、行业学会性学术组织制订的诊断标准等。诊断标准原则上要公认、先进、可行。
(2)入选标准
入选标准是指纳入的合格受试者所应具备的条件。临床试验方案应预先明确受试者入组试验的标准并在实施中严格执行。
入选标准包括:西医疾病诊断标准,有关病情与病程的分期、分型、分级的标准或规定;中医疾病与证候诊断标准;相关实验室指标和治疗情况的具体要求;对年龄、性别、婚姻状况的规定;对职业、居住地、个人嗜好状况的规定;受试者知情同意并签署知情同意书的规定等。临床试验设计时可根据临床试验目的的需要选择合理的入选标准。
(3)排除标准
排除标准是指不应该被纳入试验的各种受试者情况,其目的在于排除这些情况对于研究结论的影响。一般宜考虑下列内容:
①同时患有其他可能影响对目标适应症的诊断与疗效判断的疾病、证候或合并症者。
②已接受有关治疗,可能影响对有效性、安全性指标评价者。
③伴有可能影响疗效指标与安全性指标观测、判断的其他生理或病理状况,如月经期,或有心、脑、肝、肾及造血系统等严重原发性疾病者。
④某些可能处于高风险的人群,如孕妇、未成年人、高龄患者、过敏体质或既往有受试药物或其所含成份的不良反应史者、病情危笃而有意外事件发生可能者、或疾病的晚期患者。除非是出于临床试验目的的需要。
⑤不合作者,如不愿意接受研究措施或因患有精神等疾患不能合作者。
⑥其他:如依从性差,或因某种原因不能按期随访者。
(4)退出试验标准
分为研究者决定的退出试验和受试者自行退出试验两种情况。
研究者决定的退出:是指已经入选的受试者在试验过程中出现了不宜继续进行试验的情况,研究者决定该病例退出试验。但研究者决定该病例退出试验时,应依据预先制定的退出试验标准实施。通常,在一些危重病、可能带来不良后果的疾病的临床试验中,制定退出试验标准对于保护受试者权益,使其及时获得更有效的治疗很有必要。在制订退出试验标准时可考虑以下情况:
①病情控制情况。在临床试验中,受试者在一定时间内病情未达到某种程度的改善,以至于低于现有临床疗效,尽管尚未完成规定的疗程,但为了保护受试者,研究者可决定让该受试者退出试验,接受其他有效治疗。该病例应按“无效”病例进行统计。
②合并症、并发症及特殊生理变化情况。在临床试验期间受试者发生了某些合并症、并发症或特殊生理变化,以至于不适宜继续接受试验。
③受试者依从性情况。根据受试者在受试药物的使用、合并用药、接受随诊等方面违背临床试验方案的程度作出规定。
④在双盲的试验中破盲或需要紧急揭盲的情况。
⑤发生严重不良事件及重要不良事件等,不适宜继续接受试验的情况。
受试者自行退出试验:根据知情同意书的规定,受试者有权中途退出试验;或受试者虽未明确提出退出试验,但不再接受受试药物及检测而失访,也属于“退出”( 或称“脱落”)。受试者自行退出存在多种原因,如自觉疗效不佳,对某些不良反应感到难以耐受,因故不能继续接受临床试验,经济因素,或未说明原因等。应尽可能了解其退出的原因,并加以记录。
无论是研究者还是受试者决定退出试验的病例,应尽量追踪,尤其是因安全性原因退出试验的病例,应继续随访监测和记录受试者的转归。应保留研究病历和病例报告表,并以其最后一次的检测结果结转为最终结果,对其疗效和不良反应数据纳入全数据集分析。
(5)剔除病例标准
在分析数据时,不同的数据集应有相应的病例剔除标准。
如受试者不符合纳入标准而被误纳入试验;或符合排除标准中任一项者;或虽符合纳入标准而纳入后未曾用药者;或无任何复诊记录者;或受试者于试验期间违背方案自行换药或加用非规定范围内治疗用药,特别是合用可能影响对受试药物评价的药物,影响有效性和安全性判断者,均应考虑列入相应数据集的剔除标准。对剔除病例的判断需要在盲态审核时认定。
2.导入期
有些药物研究需要消除已经服用类似药物的延迟作用和/或稳定基线水平,因此,应设计导入(清洗、洗脱)期。导入期的长短取决于试验目的、受试药物和适应症(包括中医证候)等情况。导入期通常应停用原用药物,可以采用安慰剂导入,以排除易受安慰剂影响以及依从性差的受试者进入试验。已知半衰期的药物,通常按5个半衰期确定导入期时间。经导入期后仍然符合临床试验方案制定的入选标准时,方可开始临床试验。在导入期若病情不允许停用原用药物,可在使用相对固定的药物和剂量情况下,待病情相对稳定后,再开始临床试验。此外,有些试验需在一定时间内对某些检测指标控制、或受试者具备良好的饮食生活习惯后才能进行临床试验,也应设置相应时间的导入期。
临床试验中对照的设置常采用安慰剂对照、阳性药物对照。在剂量研究中也可采用剂量-效应对照。对照可以是平行对照,也可以是交叉对照。在中药新药临床试验中,根据具体的临床试验目的,同一个临床试验中可以采用一个或多个类型的对照。
1.安慰剂对照
安慰剂对照试验适用于以下情况:所研究疾病目前尚无已知公认有效的治疗方法;自限性疾病;某些慢性病自然病程反复波动变化,短期不治疗不至于明显影响疾病的预后;某些易受心理因素影响的疾病,具有精神症状的疾病或精神疾病;疗效判断缺少明确客观的检测指标;受试者使用常规治疗存在不能忍受的不良反应,且已证明常规治疗无效或其风险超出预先的估计。
使用安慰剂对照应符合伦理学要求,不应损害受试者健康和加重其病情。急危重症不适宜单纯应用安慰剂,可采用加载试验设计。如果受试药物和安慰剂对人体的固有反应有较大差别而使得临床试验难以保持盲态,则应采用相应的技术尽量保证试验的盲态。
2.阳性药物对照
阳性药物原则上应选用有充分临床研究证据,且当前临床普遍使用的同类药物中疗效较好的已上市药物。
所选阳性药物应该在说明书标明的适应症人群、剂量、给药途径、给药间隔、给药周期范围内使用,即其说明书适应症应与药物拟定适应症一致,且阳性药物使用的剂量、给药方案必须是该药的最优剂量和最优方案。在选择已上市中成药作为阳性药物对照时,还应考虑药物与阳性药物在功能主治、中医辨证分型上的可比性;在选择化学药品作为阳性药物对照时,在适应病种上应具有可比性。
如果使用阳性药物对照进行等效、非劣效性比较,需注意预先合理设定等效、非劣效性比较的“界值”,既要符合统计学原则,也要符合临床医学专业要求。“界值”应是临床上能接受的最大差别,并应当小于历史上阳性药物与安慰剂的优效性试验所观察到的差异;确定此差异时要考虑其变异性。对阳性药物对照的等效性试验,需指定这一“界值”的上限和下限;而对阳性药物对照的非劣效性试验只需要指定下限。
样本量的估计是临床试验设计的关键点之一。临床试验所需样本量除应满足法规最低病例数要求(如有)外,还应满足统计学的要求,以确保对试验目的给予一个可靠的回答。样本的大小通常依据试验的主要指标(疗效和或安全性终点)来确定,同时应考虑试验设计类型、比较类型等。
样本量的确定与主要指标的类型(定量指标或定性指标)、
检验假设、I类和Ⅱ类错误率、设计的类型等有关。样本量的具体计算方法以及计算过程中参数(如总体均数的差值及其变异、总体率的差值或比值、非劣效界值等)估计及其依据应在临床试验方案中列出。在确证性临床试验中,样本量的确定主要依据前期探索性临床试验结果并结合已发表的相关研究文献来保守估算所需参数,I类错误率常取单侧0.025,Ⅱ类错误率应不大于0.2,有多个主要指标或多个分组时要考虑是否对I、Ⅱ类错误率进行调整。在探索性临床试验中,如需计算样本量,所需参数的估计值可依据相关研究的结果或研究者的经验预期来估算,而I类错误率和Ⅱ类错误率则应根据试验目的进行合理的取定,可较确证性临床试验的取值适度放宽,但应满足试验目的要求。
给药方案包括临床试验给药剂量、给药方法、疗程、合并治疗的规定等。
1.给药剂量
给药剂量应根据I期临床试验耐受性及药代动力学试验结果、既往临床用药经验等进行设计。安全性也是给药剂量设计时需考虑的重要因素。Ⅱ、Ⅲ期临床试验剂量一般应低于I期的最高剂量。
2.给药方法
给药方法一般根据人体药代动力学试验结果确定,否则应根据立题依据、既往临床用药经验、拟定适应症的特点、预期药物活性等因素决定,有时也需通过临床试验研究确定。
3.疗程
临床试验的疗程是指对目标适应症所规定的药物治疗的持续时间。
应根据疾病的发展变化规律和药物临床定位、临床试验目的、作用特点确定疗程。一般需考虑:疾病的病因、病理、发生、发展及转归规律;药理毒理研究结果;文献资料及既往临床用药经验等。
早期探索性试验中,疗程设计可根据药物预期效应的起效时间和疗效最佳时间确定,同时还应考虑疾病的演变规律,尤其对于自愈性疾病,疗程设计须注意自然病程对疗效的影响。
在确证性试验中,疗程设计还应充分考虑到药物预期在临床实际使用的情况。
4.合并治疗的规定
合并治疗是受试者在临床试验期间因疾病治疗的需要所同时进行的治疗方法,包括手术治疗、药物治疗、针灸治疗等各种临床常规治疗方法。合并治疗必须预先规定,否则会严重干扰对药物有效性和安全性的评价。在方案设计时对合并治疗应考虑如下因素:
(1)在符合伦理学原则的前提下,考虑临床试验目的和适应症特点,尽量避免使用影响受试药物有效性、安全性评价的药物或治疗方法。
(2)对合并治疗的规定需符合公认的临床治疗原则。
(3)如果某些合并用药的使用是因为受试药物无效后的补救治疗,则应在临床试验设计受试者退出试验标准中预先确定,并按“无效”病例纳入分析数据集。
(4)预先规定的合并用药需考虑是否与受试药物具有药物相互作用。
(5)需对临床试验期间的所有合并用药进行详细记录,包括使用原因、使用量、使用频率等。
基线是指在随机化即刻或之前方案许可的时间窗内受试者的基础信息,包括临床试验预先设计的主要疗效指标和安全性指标的初始数据等。获取基线数据主要是为了评价组间的均衡性以及必要时进一步的分层分析。
1.基线
(1)人口统计学指标
包括出生日期、民族、性别、身高、体重等。
(2)生命体征
包括体温、脉搏、心率、呼吸、血压等。
(3)体格检查
包括常规心血管系统及神经系统检查等。
(4)既往病史:对于所有入组的受试者,都应尽量收集可能影响主要器官功能的疾病(如肾功能不全、肝功能不全、心脏疾病)的基线数据。这些数据有助于确定所纳入受试者的部分不良反应是否是由相应疾病导致的。必要时还需收集对疾病有影响的特定组织系统的病史数据,并在方案中加以描述。
(5)既往治疗史:应记录受试者的既往治疗史,并保留相关医学诊疗证据,用以辅助证明受试者符合纳入标准,或作为安全性判定的依据。
(6)疾病的诊断、分期、病情程度、病程、发病部位等:此类基线数据有助于判定疾病诊断的准确性、可靠性及受试者所处疾病发展过程中的情况等。
(7)伴随疾病和伴随治疗:如肾脏疾病、糖尿病、高脂血症等患者的伴随疾病以及伴随治疗情况。
(8)合并治疗:包括临床试验设计预先规定可使用的合并治疗方法、临床试验过程中非预先规定的合并治疗方法,如降糖药物试验中高血压病患者基线时使用的降压药物的用法、用量等。
(9)试验有效性、安全性评价关键指标的基线数据:如主要疗效指标所涉及的疾病症状、体征、量表以及重要的实验室检查值等。
(10)可能影响有效性、安全性评价的其他因素:如吸烟、饮酒等;女性受试者还应获得月经状况和末次月经周期等基线数据。
2.基线值的取得
基线值的取得通常是在随机化之前进行。基线值的取得需注意如下因素。
(1)由于大多数临床试验是在已经过治疗的受试者中进行,因此,临床试验纳入的受试者需设置导入(清洗、洗脱)期,以排除既往药物的延迟作用。
(2)目标适应症人群经生活方式干预(如饮食、运动等)仍未改善者,如高脂血症,则应在方案中详细规定基线值的确定方法(包括检测时点,检测次数等)。
(3)有些试验的基线值可能是不同时点测量值的均值,以减少不同时间点基线值的变异性,保证药物治疗前和治疗后疾病状态的稳定性。对此应进行预先的明确规定,特别是有效性和安全性评价的关键指标的基线值测量要求应在临床试验方案中作出明确规定,必要时应制定相应的标准操作规程(SOP)。
3.基线数据的分析与均衡
从理论上讲,大样本时通过随机分组的受试者组间基线是均衡的。但是由于临床试验往往受样本量的限制,在这种情况下对基线作统计描述和比较是必要的。
基线数据反映受试者在纳入临床试验最初的疾病情况,也是药物有效性和安全性评价的基准参考值。通过基线数据的统计学描述,可以判断所纳入的目标适应症人群是否符合临床试验的纳入、排除标准,是否代表目标适应症人群等。
基线数据均衡性分析的目的是检查试验组和对照组非处理因素是否一致,如果这种不一致有利于药物的疗效评价应引起重视,应仔细说明这种不一致是否导致I类错误增加。应在临床试验设计时预先设定均衡性检验的统计方法。需注意的是,如涉及有效性、安全性评价的关键基线数据出现不均衡,应采用适当的统计学方法(如分层分析或引入协变量分析模型等)进行敏感性分析。如果试验结论对不均衡的基线变量敏感,将会导致试验结果无法获得合理的解释。基线的不一致性将损失试验的检定灵敏度。对于已知的影响疗效的基线变量,可采用分层随机或动态随机化的方法进行组间均衡。
有效性指标又称为疗效指标,是反映药物作用于受试者所表现出的有效性的主要观测与评价工具。主要包括疗效观测指标和以疗效观测指标为基础用于药物疗效比较的评价指标(即疗效评价标准)。
1.疗效观测指标
疗效观测指标是用于评价药物有效性的主要观察和测量工具,可以是疾病临床终点(如死亡、残疾、功能丧失)、影响疾病进程的重要临床事件(如心肌梗死、脑卒中的发生),也可以是反映患者社会参与能力(残障)、生存能力(残疾)、临床症状和/或体征、心理状态等内容的相关量表或其他形式的定量、半定量或定性的指标;也可以是通过某些医疗仪器和设备测量手段获得的数据或检查结果,主要包括影像学、病理、生化等指标(如病理检查结果、细菌培养、血脂、血压等)。
临床结局指标是指能够反映患者的主观感觉、功能变化的特征性指标以及疾病的终点(如死亡、残疾、功能丧失)和某些重要的临床事件(如骨折)等指标。临床结局指标能直接评价药物真实的效应,如症状缓解率、疾病病死率或者临床严重事件发生率等。但由于某些疾病临床结局指标的评价往往需要的时间长、样本量大、研究成本高,有时还存在伦理学风险,导致临床结局指标观测存在困难或不合理。因此,常以易于观察和测量的疗效指标以替代临床结局指标评价药物的有效性。
替代指标是指能够替代临床结局指标、反映和预测临床结局指标变化的指标。替代指标应该是根据流行病学、治疗学、病理生理学或其他科学的证据,能够合理预测临床受益或者对临床结局指标存在疗效的指标。如血压、低密度脂蛋白作为替代指标可以预测心血管事件的发生率。需要特别注意的是,虽然替代指标可以降低药物研发成本和试验难度,但不是真正的临床结局指标,且能够广泛使用的替代指标并不多。替代指标可能因为选择不当而导致试验失败,因此,选择替代指标需要特别谨慎。有的替代指标即使已经被同类药物治疗某一疾病的临床试验验证过,但用于另一个同类适应症的中药新药仍然可能存在风险。
2.主要疗效指标和次要疗效指标
在一项临床试验设计中,疗效指标应分为主要疗效指标(主要终点)和次要疗效指标(次要终点)。
(1)主要疗效指标
主要疗效指标是反映临床试验主要目的的指标。
在确证性临床试验中,反映药物有效性的主要疗效指标一般应该是该目标适应症的临床结局指标或公认的替代指标。
主要疗效指标的选择需注意:①主要疗效指标不能随意确定,应该与药物拟定的目标适应症、临床定位和临床试验目的相一致;②主要疗效指标不宜太多,通常只有一个。但有些适应症应选择多个不同维度、相关性较低的主要疗效指标,并应考虑对I类错误进行控制;③主要疗效指标应具有较好的效度和信度并被广泛采用、容易理解;④主要疗效指标应该符合当前国内外相应适应症领域的共识。
(2)次要疗效指标
次要疗效指标是指与临床试验主要目的相关的重要支持性疗效指标,或与次要目的相关的疗效指标。次要疗效指标可以是多个。次要疗效指标可以为疗效确定提供支持,但不能作为疗效确证性依据。如与主要疗效指标相关性较强的次要疗效指标应当与主要疗效指标之间显示相应的逻辑关系。
(3)疗效评价
药物的临床有效性通过疗效观测指标来记录,疗效评价可以是某一疗效观测指标的直接测量结果,更多的是在直接测量结果基础上转化而来的、特定的评价指标来评价。因此,无论是主要疗效指标还是次要疗效指标的确定,除了需要确定疗效观测指标外,更重要的是根据临床试验目的,以疗效观测指标为基础确定疗效评价指标。同一疗效观测指标,可以转化出多种疗效评价指标,不同的疗效评价指标在药物有效性确定中的作用不同,如某一适应症的评价量表,以治疗前后的减分值为主要疗效指标,而以50%的减分率为次要疗效指标。在临床试验方案中必须预先明确设定并说明哪种疗效指标为主要疗效指标,哪种为次要疗效指标,不能在试验开始后对主要疗效指标进行期中调整,更不能在临床试验结束后再行调整。
需要特别关注的是,在某些药物的有效性评价中,为了可直观地比较临床疗效,有时把连续的计量疗效观测指标转化为分类指标,常见的是根据某一标准(截断点)转化成二分类,如“有效”、“无效”两类。如对一个连续计量疗效指标以最低改善百分率等于或超过某一阈值(如“痊愈”或“临床控制”)患者的比例作为疗效评价指标,这种疗效评价指标同样也应在临床试验设计方案中事先作出明确的规定。
一般不主张将定量指标简单地转化为多分类等级指标。因为这种转化缺乏足够科学性的基础;更不能事后随意划分截断点(如计算痊愈率、愈显率、有效率、总有效率等)进行组间比较,以免导致I类错误率无法控制。
3.中医证候
中医证候的诊断与评价可以采用量表的方法,即根据某一中医证候相关的症状体征轻重及对中医证候属性确定的贡献度进行赋分。
中医证候量表一般分为中医证候诊断用量表和中医证候评价用量表,中医证候诊断用量表和中医证候评价用量表应该分别制定,一般不能用中医证候诊断用量表甚至简单的诊断标准直接作为中医证候评价用量表。
评价中医证候变化的中医证候评价用量表应该是能够反映证候动态变化性特征的指标为主构成。
如果缺乏信度、效度评价的中医证候评价用量表,中医证候疗效可以采用减分率(最好采用消失率/复常率)按二分类资料进行统计比较分析,减分率建议根据适应症临床实际治疗情况预先设定具有临床价值的界值。
4.疗效指标观察和测量关注点
不同的疗效指标观察和测量的方法不同,同一疗效指标不同
疗效评价方法也可以有不同的观察和测量方法,因此在临床试验设计时,除了需要预先确定疗效指标外,还应该详细规定其观察和测量的具体方法,并注意测量环境、测量方法、测量质量的要求和控制等。
对稳定性较好的疗效指标,如重要临床事件的发生率等看似简单指标,也应该制定符合国内外共识的具体观测和判断的详细标准(如判断骨折愈合的标准等)。对于主观性较强或容易变异的疗效指标,有时需要在观测时间窗内(包括基线)多次测量,并规定用于疗效评价的测量值的取值要求。应该制定相应的标准化操作规范或指南。
对于量表的使用应制定相关的使用指南和标准、存在争议时的处理方法等。
5.疗效指标观测时点的设置
疗效指标的访视点一般包括基线访视点、中间访视点、试验结束访视点、随访期的访视点等。
不同的疾病,不同的试验目的、不同的疗效观测指标,其访视点的设计要求不同。
以发作性疾病的评价为例,评价控制急性发作和评价减少发作频率,其临床访视点的设置完全不同。如评价偏头痛急性发作的止痛效果,一般以开始用药后2小时内头痛及其伴随症状的缓解率作为主要疗效指标,其访视点设置应该设计为2小时。而如果是偏头痛减少发作频率的观察,多以观察一个月内偏头痛发作频率的变化为主,其访视点设置应该为一个月。
如果是评价患者一段时间内的疾病变化情况,如用匹斯堡睡眠量表评价近一个月的睡眠情况,则疗效指标访视点的设置也应以月为单位,并且基线取值也应是治疗前一个月的病情变化平均取值。此时为获得可靠的基线取值需要较长的导入期;而用于确定药物疗效的最终访视点一般不能少于几个观察时间单位,否则难以获得药物治疗后稳定可靠的疗效。
有些疗效指标的变化有其固定的生理周期和规律,如糖化血红蛋白的变化周期一般为120天左右,在选该类疗效指标进行观察时,应该以其变化周期为基础设置访视点,访视点的设置间隔也应与其变化周期基本一致。
以观察症状体征消失时间等为疗效指标时,其疗效指标访视点应该根据疾病的发病特点,在预计症状体征消失开始到结束,设置较为密集的访视点。
当某些疾病的疗程与临床试验周期不一致时,其主要疗效指标的最终访视点应为临床试验周期规定的末次访视点。
如果主要疗效指标是疾病的终点(如死亡、残疾、功能丧失)和某些重要的临床事件(如心血管事件、骨折发生),其最终访视点一般应该设置足够长。
对自限性疾病或可自行缓解的发作性疾病急性发作期的症状控制治疗为主要目的的临床试验,其主要疗效观测指标的访视点一般不宜超过疾病的自然缓解病程。如果还需要观察疾病是否出现并发症、病情复燃或疾病复发情况,应设置合理的随访期的访视点。
上市前安全性研究的目的是识别安全信号,评估安全风险,为药物风险/受益评估提供安全性数据,为上市后确定风险控制和风险最小化提供依据和方法。
中药新药在临床试验前,需依据处方组成、既往临床经验、纳入目标适应症人群特点、药理毒理研究结果,进行安全性方面的临床试验设计与实施。安全性研究还应充分重视早期临床试验所发现的问题,在后续的临床试验(特别是确证性临床试验)中及时补充和完善有针对性的、敏感的安全性观测指标。
需要指出的是,安全性指标不仅是指实验室检查指标,还应当包括所有的症状、体征等临床表现。
用于长期治疗不危及生命疾病的药物(如连续治疗6个月或以上,或间断治疗的累计时间大于6个月),暴露 6个月的受试者需要 300~600名,暴露1年的受试者需要100名。
1. 必须观察的安全性指标
(1)所有发生的不良事件(包括症状、体征等)。
(2)一般体格检查(身高、体重、体温、血压、心率、脉搏等)、重要体征检查(如神经系统等其他特殊的临床检查等)。
(3)实验室检查:
血常规检查。
肝功能相关检测指标:丙氨酸氨基转移酶(alanine aminotransferase,ALT),天冬氨酸氨基转移酶(aspartate aminotransferase, AST),总胆红素(total bilirubin,TBil;当TBil增高时,应追查直接和间接胆红素),碱性磷酸酶(alkaline phosphatase,ALP),γ-谷氨酰转肽酶(γ-glutamyl transpeptidase,GGT)。
肾功能相关检测指标:尿常规及尿沉渣镜检,微量白蛋白尿(推荐使用即刻尿白蛋白与尿肌酐的比值,UACR),血清肌酐(Scr)和/或eGFR(推荐使用简化MDRD公式或CKD-EPI公式),尿N-乙酰-β-D氨基葡萄糖苷酶(尿NAG酶)。
心脏功能相关检测指标:十二导联心电图(需常规观察ST-T改变、病理性Q波、各种心律失常、QT/QTc间期)。
2.视情况增加的安全性指标
除上述必须观察的安全性指标外,还应根据药物具体情况和目标适应症及纳入人群等特点,合理增加相关安全性指标,常见考虑如下:
(1)增加大便常规和/或大便潜血:
如受试药物可能导致消化道出血或炎症,或可能对出凝血功能有潜在影响的。
(2)根据处方药味及相关成份:
如处方中含有马钱子,应增加神经毒性等相关检查指标。
(3)根据目标适应症及纳入人群特点:
如妊娠期、哺乳期妇女用药,应增加全面的子代安全性考察内容等;更年期综合征药物,应监测相关激素水平,还应观察对乳腺、子宫内膜、卵巢的影响。
(4)由于药物药理作用可能导致的安全性影响:
如具有峻下逐水作用的药物需进行电解质等相关指标的检查。
(5)根据既往/前期临床应用经验:
既往或前期临床应用或研究中出现异常或可疑的不良反应,应增加相应的安全性指标。如前期临床应用/研究中出现可疑的皮肤紫癜,应增加相应的皮肤、黏膜观察并加强对血小板及出凝血指标的监测并增加检测时点等。
(6)根据非临床安全性研究结果:
如非临床安全性研究结果提示可能对血糖指标产生影响,需增加糖代谢等相关指标。
(7)由于证候转化可能导致的安全性问题:
如具有补益肾阳作用药物的长疗程使用可能引起补阳太过耗伤阴津导致虚火上炎的,应增加相应不良反应的观察。
(8)根据给药途径和剂型的特点:
如注射剂、眼用制剂、外用制剂等均应增加相应的与有关给药途径有关的安全性观测指标。
(9)联合用药且有安全性风险的或可能存在药物相互作用的,应增加相应的安全性指标。
(10)药理毒理研究提示对心脏可能有潜在毒性的、处方含已知具有潜在心脏毒性的、早期临床试验观察到药物可能对心脏有潜在毒性的,需增加心脏肌钙蛋白I或T(cTnI或cTnT),脑钠肽(BNP)或氨基末端脑钠肽前体(NT-proBNP)及超声心动图检查。
(11)药理毒理研究提示有肾毒性或处方中含有肾毒性报道的药物,早期临床试验观察到药物可能对肾脏有潜在毒性的,需增加血清胱抑素C( Cystatin C)、eGFR。
(12)临床试验期间视情况需增加的安全性指标:
当肝功能检查发现TBil增高时,应追查直接和间接胆红素;当肾功能检查发现尿常规检查尿蛋白阳性、且1~2周后复查仍为阳性者,增加24小时尿蛋白定量检查。尿沉渣镜检发现血尿、且48小时后复查仍有血尿者,增加尿红细胞位相检查。连续2次尿NAG酶升高2倍及以上者,增加其他肾小管功能检查指标如尿α1微球蛋白(尿α1-MG)、尿β2微球蛋白(尿β2-MG)、尿视黄醇结合蛋白(RBP)等。
3.安全性研究设计与实施要点
(1)安全性观察与检测方法的质量控制
应重视安全性指标观察与检测方法的标准化和一致性。在多中心试验中,应建立各实验室检测标准化的方法,以保证各实验室检测结果评价的一致性,并预先明确实验室指标的正常值范围。对一些关键而重要的指标,需采用中心实验室检测。需要第三方评价的安全性检测指标(如ECG、CT等)应预先规定评价方法和质量控制的具体要求。
某些易受饮食及生活方式影响的观测指标,应预先明确受试者相关饮食禁忌(如不宜高蛋白/高脂/高糖饮食,不宜饮酒/茶/咖啡)及生活方式(如不宜剧烈运动,避免劳累)等。
应针对参与安全性研究的人员(研究者、实验室检测人员等)制定相关SOP,以保证临床试验期间异常值的及时预警和有效处理。
(2)安全性指标检测时点的设置
安全性指标检测时点(包括时间窗)的设置需符合疾病发生发展变化的规律、相关指标变化规律及药物的特点等因素。
对于大于或等于6个月的长疗程药物,常规安全性指标检测间隔时间一般不应大于3个月,如为中药有效成份和有效部位制剂,常规安全性指标检测时间不应大于1个月。当存在安全性风险或前期临床试验已出现安全性风险信号的情况下,应及时增加检测时点。
心、肝、肾脏相关安全性指标检测时点具体要求详见本指导原则心脏、肝脏、肾脏安全性评价要求。
(3)实验室检测指标异常值的复查及随访
应预先制定临床试验期间或临床试验结束后实验室检测结果出现异常值的复查、随访要求,保证及时预警和采取相应措施,直到实验室检测指标恢复正常或稳定(如果不能期待完全恢复正常)。应在研究病历和病例报告表中完整记录处理过程、随访数据及转归,附上相关检查报告单。
对于因此退出研究的受试者,也应密切随访,记录转归及相关数据。
(4)严重不良事件或重要不良事件的处理及评价
应预先规定出现严重不良事件或研究者认为重要不良事件的处理措施。建议预先规定医学专业的分析和评价要求。
(5)不良事件严重程度的确定
应使用公认的标准。尤其需要比较试验组和对照组不良事件的发生率时,应在临床试验方案中预先规定。
(6)规范记录合并治疗
及时、完整、规范地记录合并治疗方法,有助于不良事件(包括实验室检测异常值)与药物因果关系的分析和判断。
如有合并用药,每一种合并用药记录应至少包括用药原因、用药开始日期、药物名称、剂量、用药次数、用药结束时间、用药结果。
4.不良事件术语及标准化
对不良事件表述相似字面术语的不正确、不一致的编码,以及对无关字面术语的不恰当的“类聚”,会扭曲安全性信号。只有预先规定并采用统一的、正确的标准术语,对不良事件进行始终如一和准确的表述,才能使发现安全性信号的机会最大化。
建议在一个中药新药的整个临床试验中采用统一、标准的不良反应编码惯例或字典(如MedDRA等)。临床试验前预先选择一种公认的标准字典,并形成相应的数据库,数据管理应建立标准流程,且应适时更新字典并保证医学和药物编码在不同版本字典之间的一致性。临床试验使用的字典版本应储存在数据库里。中医药领域特有的不良事件需按相关程序和要求及时补充纳入所采用的字典。
如果预先没有使用统一的不良事件编码,在分析和总结不良事件时,既要澄清研究者开始使用的术语,也要对有关不良事件进行归类( 相关不良事件可能代表同一现象),以保证明确真正的不良事件发生率。研究者开始使用的和后来经改动的术语均应说明。
5.安全性数据集的构成
(1)安全性数据集的定义
只要使用过一次药物的受试者均应列入安全性数据集。
应对退出/脱落病例的原因进行查实和确认,包括认为缺乏疗效、失访、撤除知情同意书或者其他原因等,应尽量查明导致脱落或退出的各种具体的原因,不能简单记录含糊解释,如“撤除知情同意”“失访”“未回访”等,还应尽可能地全面收集其退出时的安全性数据。
应鼓励退出的受试者说明原因并提供有关信息,尤其需要关注是否为安全性问题。
有些受试者退出原因为实验室数值异常、轻微的症状或体征变化等,虽然尚未达到规定的不良事件程度,也应收集其信息。
(2)安全性数据的汇总
在临床试验安全性数据中,任何实验室参数变化及生命体征、体格检查异常都可能提示药物的潜在毒性或风险。因此,在临床试验总结报告中应对受试药物和对照药物(包括安慰剂)的所有不良事件进行详细的描述和报告,包括临床症状、生命体征和实验室检查结果的异常改变等。
对不良事件的汇总应包括以下内容:
通过表格和图形方式汇总所有不良事件;对每例不良事件逐例表述;严重或重要的不良事件还应逐例进行详细报告和分析。
如果数据较多,还应按归类 (如血液学、肝功能、ECG等)分为多个表格。同一个指标,可设计多个表格,从不同角度进行分析和比较。表格应根据中心和组别分组,还应根据是否与药物有关来区分。应明确受试药物及对照药物的组别。
①不良事件
每例不良事件的表述至少应包括如下内容:
受试者编号,中心编号
年龄、性别
不良事件表现
不良事件发生时间
不良事件发生的时间与最近1次给药之间的间隔时间
严重程度(轻、中、重度)
危及生命性(危重的或非危重的)
不良事件持续时间/发生频次
处理措施(无,减量,停止治疗,其他特殊治疗,如药
物名称、剂量、次数等)
转归
合并治疗情况
因果关系判定(如何判断应在表中或其他地方说明)
②严重不良事件和重要不良事件
针对严重不良事件和研究者认为需要报告的重要不良事件应逐例进行汇总,并在临床试验总结报告附件中提供原始门诊病历或住院病历资料。在按照不良事件汇总基础上,还应包含如下内容:
既往病史
和不良事件有关的联合用药的起始日期
有关的体格检查结果
有关的检查结果(如实验室、ECG、病理检查结果等)
接受过的治疗
再次用药结果(如有)
结局和随访信息
诊断结论(附专家讨论会,会诊记录等)
③涉及肝损伤相关不良事件:根据肝损伤病例报告表的要求汇总,详见肝脏安全性评价要求部分。
6.安全性数据的分析
对安全性数据分析应从三个方面考虑:
首先,需根据确定暴露的程度(剂量、用药时间、样本量),以决定可在多大程度上评价安全性。
其次,应针对常见的不良事件、实验室检查指标的异常进行确认并判断与药物的相关性,进行合理的分类,在试验组、对照组(和/或安慰剂组)以不良事件或不良反应发生率之间进行比较和分析。
最后,针对严重不良事件及重要不良事件、退出试验病例及死亡病例的分析,以发现危重的不良反应和其他重要不良反应。
(1)药物暴露的程度
通常包括药物暴露的样本量、暴露持续时间和暴露的剂量。
暴露持续时间:以药物使用时间的平均数或中位数来表示。
暴露剂量:以药物剂量的中位数或平均数来表示,也可以表示为每日平均剂量下有多少受试者数,或常规剂量、最大剂量下有一定暴露时间的受试者数量。
暴露持续时间和暴露剂量也可以根据年龄、性别以及疾病的严重程度等分层表示。
(2)不良事件的描述和分析
描述所发生的不良事件最简单的表达方法是粗率(crude rate),粗率是根据发生不良事件的患者数除以接受药物治疗的患者总数。
对每一例不良事件应按轻、中、重度程度归类,并根据身体系统进行归类。应进行组间比较,统计每组出现不良事件的受试者例数及发生率,结合不良事件的严重程度(采用公认的毒性分级标准)及因果判断分类进行。
对每项实验室指标检查值及生命体征、体格检查指标应进行统计学描述。并提供试验前正常/试验后异常、试验前异常/试验后异常加重等的汇总情况。
临床试验出现的所有不良事件均需进行与药物相关性的分析,明确与药物相关的不良反应、尚不能排除与药物相关的不良事件。
(3)严重不良事件、重要不良事件的描述和分析
对严重不良事件和研究者认为的重要不良事件需进行逐例分析,以确定/排除是否属于药物严重或重要的不良反应。
应明确严重不良事件、重要不良事件的性质和严重程度,导致严重不良事件、重要不良事件发生的临床过程及与治疗的相关性,如剂量、药物浓度、合并用药、受试者其他情况等(如既往疾病史、既往治疗史),进行详细的分析并明确与药物的相关性。
(4)退出试验病例及死亡病例的分析
对因不良事件(不管是否与药物有关)而退出试验或已死亡的受试者进行分析,以发现严重的不良反应和其他重要的不良反应。
(5)识别和评估风险因素
通过临床试验安全性数据的汇总,对不良反应/不良事件的表现和发生规律进行分析,如时间依赖性、人口统计学因素(如年龄、性别、种族等)、与剂量、药物浓度和给药方式的相关性、基础疾病情况(如肾功能异常)等,寻找可能影响不良反应/不良事件发生频率的因素,明确与药物相关的不良反应及尚不能排除与药物相关的不良事件,以评估药物安全性风险发生的原因(高风险因素和高风险发生人群)。
在安全性评价中,还需根据临床试验所有安全性数据,提出相应的风险控制/风险最小化的方法,为药物的风险/受益评估提供依据。通常风险控制/风险最小化的方法包括在拟定的说明书中描述药品安全、有效使用的条件,需关注的临床相关信息等,以尽量减少药物上市后临床使用过程中可能存在的风险。
(6)安全性数据报告分析与评价重点
应对各期临床试验过程中出现的全部不良事件、严重不良事件、重要不良事件进行合理的因果判断,以不良反应类型和发生率等作为临床安全性评价的基础,根据不良反应类型和发生率评价药物不良反应的严重程度。
在安全性评价中主要关注安全性数据是否完整充分有无遗漏,发生的风险是否与对照组进行了合理的比较,是否包括少见的、严重的及与剂量相关的不良反应,是否存在同类药物的安全性问题等。
7.不良事件与药物因果关系的判定方法
国际上有多种方法判断药物不良事件/不良反应的因果关系,分析其关联性。对于药物上市前临床试验不良事件/不良反应的因果关系,建议依据以下五个原则分析判定:
(1)开始用药的时间与不良事件/不良反应出现的时间有无合理的先后关系;
(2)可疑ADR是否符合该药品已知ADR类型;
(3)所怀疑的ADR是否可以用患者的病理情况、合并用药、合并治疗方法或曾用治疗方法来解释;
(4)停药或降低剂量可疑的ADR是否减轻或消失;
(5)再次使用可疑药品后是否再次出现同样反应。
关联性评价:依据以上五条原则将关联性评价分为“肯定”“很可能”“可能”“可疑”“无关”5级。
其中将“肯定”“很可能”“可能”“可疑”情况合计作为不良反应发生率计算时的分子,分母则为用于评价安全性的全部受试者例数。
值得注意的是,不良事件与药物相关性的判定为研究者个体判断的结果。当主要研究者在进行临床试验总结汇总安全性数据时,还需根据处方组成、非临床安全性研究结果、不良事件发生的频次、严重程度、趋势,对照组的不良事件发生情况等进行整体判定。
随访是指临床试验观察周期结束后,继续对受试者进行追踪访视至终点。一项临床试验是否需要设定随访要求,应根据药物作用特点、适应症特点和试验目的确定。
根据药物的不同作用特点和试验目的,随访内容包括远期疗效、疗效的稳定性、控制疾病复发作用、生存率及生存时间、迟发或蓄积的不良反应和其他安全性指标等。随访的期限与次数、间隔时间,均应根据研究疾病的自然史和对随访终点的要求等而制定。
随访可以针对进入试验的所有受试者,也可以合理选择部分病例。如随访目的是观察疗效的稳定性及疾病复发情况,可对临床疗效为痊愈、显效、有效的病例进行随访。
1.随访要求
(1)临床试验方案应对随访计划及其相应观察指标进行预先规定,研究病历和病例报告表中应有随访内容的项目。
(2)随访应尽量在盲态下进行,以避免偏倚。
(3)按照试验方案中的随访要求观察需要的临床结局。
(4)根据试验目的,确定随访人群范围。
(5)尽量采用客观的随访检测指标。
2.随访指标
根据随访目的选择相关的随访指标,通常有以下项目:
(1)远期疗效;
(2)疾病复发情况;
(3)安全性观测指标,如发生不良事件时,应随访至完全正常或稳定(医学上认为可以停止观察)为止;
(4)临床症状和客观指标变化情况,如体检、X线照片、ECG和实验室其他检查等;
(5)死亡和死亡原因;
(6)生存质量(在临床试验中特指与疾病相关的生存质量)。
3.随访结果的评价
评价随访结果的指标有治愈率、缓解率、复发率、病死率、生存(存活)率等,应根据临床试验目的合理选择。
临床试验及随访期间出现不良事件者,应评价不良事件的转归及分析原因,并纳入安全性数据集分析。
随访期间受试者病情、使用药物情况发生变化,应客观报告随访结果,并作出统计学分析。
(十一)试验的中止
试验中止是指整个临床试验尚未按照试验方案完成即中途停止。试验中止的目的主要是为了保护受试者权益,保证试验质量,并避免不必要的经济损失。
临床试验的中止原因通常有以下方面:
1.试验中发生严重安全性问题,研究者认为受试者的安全性可能受到损害时;
2.试验中发现药物治疗效果太差甚至无效,不具有临床价值;
3.在试验中发现临床试验方案有重大失误,或在实施中发生了重要偏差,难以评价药物疗效和/或安全性;
4.申请人提出中止(如经费原因、管理原因等);
5.药品监督管理部门要求中止。
八、中药新药临床试验质量控制
良好的质量控制是保障临床试验获得可评价的有效性、安全性数据的必要条件。对可能影响临床试验质量的问题,应在临床试验方案设计时预先考虑,并在临床试验实施前采取相关措施。需要注意在早期试验中发现的影响临床试验质量的问题,如研究者评价的一致性、不同实验室检测指标一致性等,应在确证性临床试验之前进行归纳,并在设计和实施中采取相应的措施以确保临床试验的质量。相应措施应在各期临床试验方案和相关文件中预先规定和说明,实施过程的相关文件也应保存。
需关注的影响质量控制的常见因素如下:
(一)主观症状评价或量表应用的质量控制
在中药新药临床试验中,采用与疾病相关的症状、体征或量表是有效性研究的重要部分。常见的问题是不同研究者评价的一致性差,尤其作为主要疗效指标时,将影响对有效性的客观评价。在设计临床试验方案时,应采用信度、效度和反应度良好的量表和/或行业公认的症状量化标准。在使用症状、体征或量表评价有效性时,要重视对研究者评价一致性的质量控制,尤其是在多中心试验时,在临床试验实施前应对所有研究者进行统一培训,并应通过一致性检测。如应用某些特殊量表(如精神适应症领域),应请具有资质的研究者进行,或培训研究者取得资质,以保证研究者对量表使用的质量。在早期探索性临床试验中即应重视和关注研究者对量表及症状等评价的一致性,以确保进入确证性临床试验前研究者对量表及症状等所采集的临床试验数据具有可评价性。
(二)实验室检测指标的质量控制
参与临床试验的医疗机构临床检验实验室应当建立质量管理标准和标准操作规范,以保证检测、诊断数据及结果的准确可靠。鼓励采用通过卫生部临床检验中心的室间质量控制评价或通过ISO 15189认证的实验室。
需要关注多中心试验的各实验室对主要诊断指标或疗效指标一致性的质量控制。如多中心实验室检测设备、主要检测指标参考值范围不同,由于系统偏倚导致不同中心的检查结果难以合并统计分析,将影响对药物的安全性和有效性的准确评价。
如实验室检测指标为有效性或安全性评价的关键指标,或特殊的检查指标及变异性较大的指标,不同中心的实验室检查结果受检测仪器、检测条件及检查人员的影响较大,建议采用国内外权威机构认证的中心实验室一个批次检测。中心实验室需要制定严格的样本采集、储存、配送的SOP,以保证样本采集和运送的质量和安全性。确证性临床试验有效性、安全性关键指标生物样本的保留需符合相关要求。
若未使用中心实验室,由于设备、分析者和参考值范围的不同,不同的研究中心可能获得不同的实验室检测结果,此时需要采取措施取得一致性的数值,如进行检验方法统一培训和一致性测定,这对于以实验室指标作为主要指标时尤为重要。否则,有可能由于系统偏倚导致不同研究中心的实验室检查结果难以合并,从而影响对药物的安全性和有效性进行准确的评价。
(三)非实验室检查指标的质量控制
对于非实验室检查指标,如血压检查、心电图运动平板试验、X线检查、B超、CT、MRI等,应选择公认的、质量可控的测量方法和测量仪器,对检查过程要制定规范的SOP,以保证不同中心、不同人员检查测量结果的一致性。为此,需要明确对测定仪器的要求和对测量人员的技术培训要求,并在临床试验方案中,预先规定公认的、合理的技术要求、规范和标准。如测量原发性骨质疏松症的骨密度,应预先规定骨密度仪精确性误差测量规范。如果检查指标为疗效和/或安全性的关键内容,建议采取盲法评价和/或第三方评价。
(四)受试者选择及疗效评价的质量控制
在中药新药临床试验中,由于症状、体征或量表等主观指标较多,更应该重视通过合理的试验设计控制主观性偏倚。主观性偏倚是指临床试验中由于受试者和研究者对药物、治疗措施先入为主的信赖或怀疑所造成的偏倚。这是干扰药物临床试验的重要因素之一。
随机化和盲法是控制受试者选择偏倚和疗效评价偏倚的主要措施。研究者应当遵循试验方案的随机化程序,按其要求顺序进行随机化分配,保证随机化方法不被破坏。应注意盲法切实可靠的实施。由于中药安慰剂制作存在实际困难,故应加强安慰剂的研究,避免因安慰剂制作质量的问题,导致临床试验实施中可能破盲。
(五)临床试验原始数据采集的质量控制
通过临床试验原始数据的完整采集记录,可以了解影响临床试验质量控制的相关因素也有助于解释临床试验数据中发现的问题。
1.基线数据的获得:临床试验的基线筛选方法可以保证纳入符合要求的受试者,应保留相关筛选记录,如纳入标准要求以初次诊断经过生活方式干预后的目标适应症人群,则应保留生活方式干预的记录和筛选过程相关的检查记录。
2.纳入目标适应症人群的证据:在临床试验中应保留证明所纳入目标适应症人群特征的原始记录,如病史、病程、既往用药情况,重要的疾病诊断依据(如CT、B超、ECG、冠脉造影、病理报告等)。通过研究病历的设计,提醒研究者收集和整理重要的纳入目标适应症人群信息,以确保所纳入目标适应症人群符合设计的要求。
3.缺失数据的追踪:临床试验数据可能因为患者拒绝继续参与试验、不良事件的发生、患者失访等原因导致数据缺失。过多的缺失数据既影响有效性和安全性的评价、影响试验组与对照组之间的可比性,也影响所纳入目标适应症人群特征的代表性。因此,临床试验应预先制定相关措施,以尽量避免数据缺失,尽量追踪和记录所有临床试验数据的信息。
4.合并治疗的记录:在临床试验实施过程中,除预先规定的合并治疗方案外,有时难以避免使用了规定外的治疗,均应如实详细记录,并客观评估其对有效性和安全性的影响。
九、中药新药临床试验用安慰剂研制的要求
申请人应对临床试验用安慰剂的质量负责,并应保留临床试验用受试药物、阳性药物、安慰剂的样品。
安慰剂制备应符合如下要求:
1.安慰剂应不会对人体健康产生危害,不会产生明显不良反应(与受试药物相同的药用辅料产生的轻微不良反应除外)。
2.安慰剂应与受试药物/阳性药物相似,如口服制剂安慰剂应在颜色、气味、味道、形状、质感等特征方面与受试药物/阳性药物相似,使临床试验参与者难以区分。需采用合理的方法对其相似性和适用性进行判断和评价(如借鉴食品或化妆品等的感官评价方法)。
3.安慰剂对受试药物的适应症应无明显治疗作用,不会干扰对受试药物有效性的观察。
4.安慰剂所用原辅料应符合相应给药途径的质量要求。
5.应明确安慰剂处方用原辅料的种类及用量。应尽可能少加可能对受试药物安全性及有效性产生干扰的物质。安慰剂用辅料应尽可能与受试药物相同。
6.应建立安慰剂的质量标准,保证安慰剂的安全、稳定、均一。
7.安慰剂的规格、外观、包装、标签、标识等应与受试药物/阳性药物一致。
在某些特定情况下,安慰剂也可采用已上市药品(如生理盐水等),但应符合上述要求。
对于特殊给药途径用安慰剂(如注射给药、眼用制剂、外用制剂等),还需预先进行必要的相关研究以保证用药的安全性。
十、肝脏安全性评价要求
在中药新药临床试验中,药物是否可导致肝脏损害是安全性评价的重要内容之一。在现阶段对于药物性肝损伤发生机制、诊断、严重程度等认识的基础上,本指导原则修订了肝脏安全性评价指标、评价要点等具体要求,以期科学合理地评价药物对肝脏安全性的影响。
药物性肝损伤(drug induced liver injury,DILI) 是指在正常治疗或临床试验剂量范围内的药物使用过程中,因药物本身或其代谢产物引起的程度不同的直接或间接的肝脏损害。
(一)药物性肝损伤的发生机制
DILI的发生分为可预测性和不可预测性,前者通常与药物剂量相关,后者多为特异质性。目前多认为DILI的发病机制与药物及其代谢产物的直接肝毒性及机体特定的基因多态性相关。
(二)药物性肝损伤的诊断
DILI的诊断主要是一个排除性诊断过程,即使依靠肝活检也难以确诊。目前国内外有多种半定量的DILI诊断标准,其中由国际医学科学组织委员会(Council for International Organizations of Medical Sciences,CIOMS)制定的RUCAM (Roussel Uclaf Causality Assessment Method ,Roussel Uclaf 因果关系评估法)评分表(见附录),较为广泛地得到肝病学专家的认可。其主要参数是:用药、停药与发病的关系,风险因素(年龄、酒精、怀孕),其他肝损伤因素的排除,合并用药,对当前潜在肝毒性药物的认识水平和激发试验的结果。
DILI有急慢性之分,急性DILI是最常见的发病形式,占90%以上。
药物导致的急性DILI分为肝细胞损伤型、胆汁淤积型和混合型。
1.肝细胞损伤型: ALT≥3×正常值上限(upper limit of normal,ULN)且R≥5[R值为ALT实测值相对于正常值上限的倍数与ALP(AKP)实测值相对于正常值上限倍数的比值];
2.胆汁淤积型:ALP≥2×ULN且R≤2;
3.混合型:ALT ≥3×ULN,ALP≥2×ULN且2 < R< 5。
(三)药物性肝损伤的严重程度分级
通常将急性DILI的严重程度分为5级:
1级(轻度):仅肝酶增高,大多数患者适应。患者血清转氨酶(aminotransferase)或ALP水平升高,但TBil<2.5 mg/dl(42.75μmol/L),这种变化为可恢复性,并且无凝血功能异常(国际标准化比值,international normalized ratio,INR<1.5)。又可分为有症状(S)和无症状(A)2组,DILI症候群主要表现为疲乏、恶心、右上腹疼痛、瘙痒、皮疹、黄疸、虚弱、厌食或体重减轻。
2级(中度):患者肝细胞功能轻度减退。转氨酶或ALP水平升高,且TBil≥2.5 mg/dl(42.75μmol/L)或虽无高胆红素血症但存在凝血功能异常(INR≥1.5)。
3级(中至重度):患者血清ALT、ALP、胆红素或INR升高,且因DILI而需要住院治疗或原已住院的患者住院时间延长。
4级(重度):患者血清转氨酶和/或ALP水平升高,TBil≥2.5 mg/dl(42.75μmol/L),且至少出现下述情况之一:⑴有肝功能衰竭的表现(INR≥1.5,腹水或肝性脑病);⑵出现与DILI事件相关的其他器官(如肾或肺)功能衰竭。
5级(致命):患者因DILI死亡或需接受肝移植。
(四)肝脏安全性评价指标
评价DILI的肝脏血液生化学安全性指标至少应该同时包括ALT、AST、TBil、ALP及GGT(γ-GT)五项。
当TBil增高时,应追查直接和间接胆红素水平,以鉴别黄疸的性质。
当临床试验疗程超过6个月、试验过程中出现肝损伤加重、怀疑慢性肝损伤或肝功能衰竭等情况时,需要检测前白蛋白、白蛋白、凝血酶原时间、INR以及总胆固醇等指标,必要时行肝活检组织病理学检查。
(五)药物性肝损伤的评价要点
1.严重药物性肝损伤的预警信号
美国学者海曼–齐默曼(Hyman Joseph Zimmerman)提出:药物引发的肝细胞损伤如果同时伴有黄疸,为严重肝病的标志,预后不良,其急性肝功能衰竭的死亡率(肝移植)可高达10%~50% ,此现象称为海氏(Hy's)法则。该法则将TBil>2×ULN、ALT>3×ULN视作严重肝损伤的标志。在临床试验中发现1例海氏法则病例具有预示作用,发现2例可高度预示发生严重DILI的可能性。
具体来说,与(无肝毒性的)对照药物或安慰剂相比,药物引起ALT或AST升高至≥3×ULN或更多,应当引起充分注意;
如果转氨酶峰值高达10×ULN,尤其>1000IU/L者,应当引起严重警惕;
当转氨酶和TBil联合升高(转氨酶通常远高于3×ULN),在未发现胆汁淤积的情况下(ALP<2×ULN),TBil>2×ULN,这是药物引起严重DILI潜在可能的最明确、最特异的预测指标,表明受试者整体肝功能降低。
在应用海氏法则时应注意以下几点:(1)黄疸必须是肝细胞性的,而不是淤胆型的;(2)需排除急性病毒性肝炎或其他肝病;(3)引起肝损伤的药物需有可造成轻度肝损伤的证据。
除了胆红素和ALT增高外,凝血酶原时间(或INR)和血清白蛋白水平也可列入评估肝损伤程度的指标。
2.肝功能检测的时点
DILI的发生存在潜伏期,尤其是胆汁淤积型肝损伤的潜伏期较长,一般肝脏生化学检测正常的受试者,若药物的疗程≤2周,至少需要在治疗前、后各进行1次检测;疗程>2周的受试者应在治疗前、治疗开始后2周各进行1次检测,此后至少应每4周进行1次,时间应持续12周以上。如果在合理的暴露期间(如12周)后无肝损伤的表现,检测间隔可适当延长至每12周1次直至疗程结束。但是对于药物重复给药毒性研究提示或组方中含有既往有肝毒性报道的药物以及受试者为易发生肝损伤的人群等,应酌情增加检测频度。
对于基线测量值正常的受试者在接受受试药物后,如转氨酶升高超过3×ULN,或者基线测量值异常的受试者在接受受试药物后,转氨酶升高超过基线值的2倍,则应加以密切观察,以确定肝脏生化学异常是一过性并能自行缓解改善的,还是呈继续恶化的。密切观察的时间密度为每周进行肝脏生化学检测2~3次,并根据需要增加评价肝功能的检测项目。如果异常状况稳定或者研究药物已停用且受试者无症状,则重复检查可减为每周1次。对于转氨酶<3×ULN和/或TBil<2×ULN的轻度肝脏生化学检查异常者,应每周监测1次,并加强观察(详见病例报告表应收集的内容)。
DILI除了肝功能异常外,也可能发生相关的症状和体征,如食欲减退、恶心、疲劳、右上腹不适、呕吐以及发热、皮疹、瘙痒等。在某些情况下,DILI症候群的症状可能为肝损伤的早期信号。一旦受试者出现无其他原因可以解释的相关症状体征时,尽管尚未到达下一个肝功能检测的访视窗口,也应立即进行肝功能以及相关实验室检测。
(六)决定停药的判断依据及处理
一般来说,如果出现下列情况之一,应停止用药,直至肝损伤完全消除或至基线状态,同时排除受试药物所致DILI外的其他原因所致的肝损伤。
1.ALT或AST>8×ULN;
2.ALT或AST>5×ULN,持续超过2周;
3.ALT或AST>3×ULN并且TBil>2×ULN 或INR>1.5倍;
4.ALT或AST>3×ULN,并有严重疲劳、恶心、呕吐、右上腹痛或压痛、发热、皮疹及/或嗜酸性粒细胞增加(>5%)。
若肝损伤进一步加重,应及时组织肝脏病专家对受试者进行救治。
(七)随访
所有可能发生DILI的受试者均应随访至所有异常值恢复正常或至基线状态,结果应记录在研究病历、病例报告表和数据库中。
应该注意的是,更长时间的随访有时会揭示与基础肝病相关的肝损伤。
(八)再暴露
一般对需要停药的受试者原则上不应尝试进行再暴露。
出于伦理学和安全性考虑,除非药物对受试者来说获益很大而且没有可替代的治疗,或者受试药物的真实累积数据未显示可致严重肝损伤,经相关的学术委员会及伦理委员会讨论同意后,方可进行再暴露。再暴露时,必须将潜在的风险充分告知受试者并获得其同意,并对其进行密切观察和随访。对接受再暴露的受试者需进行单独的临床试验,单独制定相应的临床试验方案。
(九)肝损伤病例应收集的内容
肝损伤病例的研究病历和病例报告表应收集关于临床症状和体征、实验室异常及任何肝病的潜在原因,包含但不限于下述内容:
1.开始给药至开始发病的时间和日期;
2.停止给药的时间和日期;
3.药物再暴露的时间和日期;
4.合并用药包括非处方用药剂量、开始和结束时间、药物是否已知有肝毒性;
5.可疑药物的再暴露和去激发信息,包括开始和结束时间及剂量详情;
6.临床症状(疲劳、虚弱、恶心、厌食、腹痛、深色尿、瘙痒)和体征(如发热、黄疸、皮疹、脑病)出现及结束时间;
7.与肝功能异常有关的病史:包括存在缺血/低血压、严重低氧血症或充血性心力衰竭、败血症及怀孕等;
8.饮酒史:平均每天或每周饮酒量(应折算成酒精克数)及持续时间等;
9.潜在的肝病史;
10.肝外特点:是否存在超敏反应(嗜酸性粒细胞增多,自身抗体阳性);
11.实验室动态评估:ALT、AST、ALP、GGT及胆红素(直接和间接)开始异常、峰值以及恢复至正常的时间(注明日期);
12.病毒学指标:anti-HAV-IgM、HBsAg、anti-HCV、anti-HEV-IgM、EB病毒外壳抗原IgM抗体及巨细胞病毒IgM抗体等;
13.血液学指标:嗜酸性粒细胞、淋巴细胞及凝血功能测定等;
14.免疫学指标:免疫球蛋白、蛋白电泳、类风湿因子及自身免疫性肝病抗体等;
15.能排除其他相关肝损伤的测试;
16.影像学与组织病理学:肝脏超声、CT(computed tomography)、磁共振成像(magnetic resonance imaging,MRI)、磁共振胰胆管成像(magnetic resonance cholangiopancreatography,MRCP)或其他,必要时肝活检。
所有可能符合海氏法则的病例均应作为药物相关严重不良事件进行处理,并及时报告药品监督管理部门及伦理委员会。报告应包含所有可用信息,并应开始密切随访直至问题完全解决。
(十)药物性肝损伤与“肝脏生化学检查异常”的区别
CIOMS将肝脏生化学检查异常区分为两种情况,一种是“肝损伤”,一种是“肝脏生化学检查异常”,认为“肝脏生化学检查异常”不属于“肝损伤”。“肝损伤”是指在缺乏组织学检查的情况下,ALT或结合胆红素(conjugated bilirubin)≥2×ULN,或AST、ALP和TBil三者均升高,且其中之一≥2×ULN。“肝脏生化学检查异常”是指AST或ALP或TBil中某一项单项指标升高≥2×ULN,或ALT、AST、ALP和TBil的升高介于1~2×ULN之间。据此,受试者在接受药物后,如实验室检查显示为“肝损伤”,则要考虑到DILI的可能性;如实验室检查显示为“肝脏生化学检查异常”,则似乎暂时还够不上DILI的诊断条件,对此需要跟踪观察和随访。
十一、心脏安全性评价要求
在中药新药临床试验中应关注药物对心脏损伤的观察和评价。药物性心脏损伤是指在正常治疗或临床试验剂量范围内的药物在使用过程中出现的、与用药目的无关的、直接或间接的心脏损害,包括心律失常、心肌缺血、心脏收缩或舒张功能异常甚至心肌肥厚或心脏扩大等心脏病变。
(一)中药导致相关药物性心脏损伤的可能原因
1.某些中药的直接毒性作用可损害心肌兴奋-收缩偶联,影响线粒体的氧化磷酸化,从而影响心肌收缩和舒张功能。
2.某些中药可能通过作用于离子通道,或影响心脏起搏、传导系统,引起不同类型的心律失常。尤其多见于具有一定易感因素的患者,如老年、心衰、电解质紊乱、缺氧、心肌缺血等患者。
3.某些中药在特定人群中可能表现出心脏冠状动脉收缩作用以及增加血小板聚集,促使血栓形成,从而加重心肌缺血,导致冠状动脉粥样硬化性心脏病病情加重。
4.某些中药与化学药品(如洋地黄类)存在药物相互作用,影响药物代谢过程及血药浓度,致药物蓄积过量而引起药物性心脏损伤。
(二)药物性心脏损伤的诊断
1.明确药物与不良反应之间的关系。包括评价受试药物的药理特性、合理剂量、疗程,心脏损伤与服药之间的时间关系,停药后心脏损害恢复情况等。
2.患者自身情况。是否为易损人群,如老年、心肌缺血、心力衰竭、电解质紊乱、肝肾功能不全者等,合并用药情况(如延长QT/QTc药物)等。
3.心血管相关临床表现。(1)症状:胸痛、胸闷、心悸、呼吸困难、重度乏力、晕厥等;(2)体征:血压异常升高或降低,口唇紫绀,颈静脉充盈,脉率及心率增快、减慢或节律紊乱,呼吸急促或微弱,呼吸音异常,出现啰音,心音增强或减弱,出现异常心音或附加音、心脏杂音,下肢水肿;(3)实验室检查异常:心肌标记物、ECG、超声心动图等。
4.需除外原发和其他继发心脏疾病以及心脏外疾病。
(三)药物性心脏损伤程度的评价标准
药物性心脏损伤的程度分级评定可参考纽约心脏协会(NYHA)关于心脏功能的评估或不良事件常用术语标准(CTCAE 4.0)。
(四)心脏安全性评价指标
1.常规心脏安全性评价指标
(1)生命体征:心率、血压、呼吸频率
心率、血压、呼吸频率是评价心脏安全性最重要的观察指标,尤其血压异常升高或降低,心率、呼吸频率明显增快、减慢或节律紊乱,均应引起重视。
(2)十二导联心电图
十二导联心电图可作为常规心脏安全性评价的检查指标。ECG正常的病例暴露于受试药物后出现心电异常,或原有轻微心电异常暴露于受试药物后加重的病例,都应予以关注,包括ST-T改变,病理性Q波,各种心律失常,如窦性心动过速、心动过缓、室性早搏、房性早搏、心房颤动、室上性及室性心动过速以及各种传导阻滞等。此外,QT/QTc应是ECG检查的必测内容。QT间期即QRS波群的起点到T波终点的时程,QTc间期是指排除了心率影响的校正的QT间期,计算方法为QTc = QT/(RR^0.5),其中RR为标准化的心率值,根据60除以心率而得。QTc测量通常选择胸前导联或II导联为宜,一般以QTc延长至>450ms或较基线增加>30ms为异常标准。
检测时点:
疗程≤1周:至少治疗前、后各1次;
疗程>1周:治疗前、治疗开始后第1周,此后至少每4周1次,12周后至少每12周1次,直至疗程结束。
2.视情况增加的心脏安全性评价指标
(1)对于药理毒理研究及早期临床试验结果提示有潜在心脏毒性的药物,可考虑加做cTnI或cTnT;如前期研究提示对心功能有潜在影响者,可再加做BNP或 NT-proBNP,检测频度同上。
(2)在临床试验访视时如出现无法解释的ECG异常(例如新出现室性早搏,或室性早搏较入组时加重),特别是处方中含有既往报道有潜在心脏毒性的中药时,可考虑加做cTnI或cTnT,必要时酌情进行动态心电图和/或超声心动图检查,以明确是否有药物性心脏损伤。
(3)对于药理毒理研究及早期临床试验结果提示有潜在延长QT/QTc作用的药物,应在目标适应症人群中充分观察药物对QT/QTc的影响,除一般时点的QT/QTc检测外,尤其要关注剂量效应关系,增加药物峰效应时点的ECG检查,并加强对药物性心脏损伤易损人群的监测和不良事件(如QT/QTc>500ms、出现Tdp等严重心律失常等)的记录。
(五)心脏安全性评价时的注意事项
需要说明的是,以上部分指标改变是非特异性的,有些与检测方法、操作流程也有一定关系。因此,在观察药物性心脏损伤时,特别要注意各临床试验参加单位、不同研究者检测方法的一致性,对异常ECG的读取和判定最好由经验丰富的研究者统一进行诊断。诊断应注意给药前后的检测结果对比,分析可能的合并用药影响,并结合病史、临床症状、体征进行综合判断。
(六)受试者安全的保护措施
1.对非临床研究显示有潜在心脏毒性的,在制订I、Ⅱ期/早期试验方案时,应重点监测药物相关的心脏不良事件,注意其与非临床研究结果的一致性及其原因,剂型和给药方式的合理性(如胃肠道外给药是否增加不良反应)。临床前研究及Ⅰ、Ⅱ期/早期临床试验如观察到药物对心脏有潜在影响的,应结合药物的临床适应症及风险/受益进行综合评估,以确定是否有必要进行后续的临床试验。在后期(尤其是确证性)临床试验方案设计中,应充分考虑纳入人群、必要的监测指标及受试者保护措施。此类中药新药的临床试验设计与实施过程中,不论何种适应症,均应有心脏内科专家参与。
2.在临床试验实施过程中,应预先告知患者需监测的症状,如心慌、胸闷、胸痛、喘憋、呼吸困难、重度乏力等,一旦发生以上不适,即使未到访视时间,也需及时联系研究者。研究者应酌情进行必要的ECG、cTnI或cTnT、BNP或NT-proBNP、超声心动图、动态心电图等理化检查,并给予相应的对症处理,详细记录不适发生的时间、诱因、症状特点及药物服用情况。如果出现检测结果异常,不论有无临床症状,都应结合患者具体情况判断与药物的相关性及其临床意义,酌情予以停药退出试验或继续密切观察,定期随访直至异常指标恢复正常。
十二、肾脏安全性评价要求
肾脏是体内药物代谢和排泄的重要器官,由于其自身解剖与生理特点,容易受到各种药物的损害。中药相关肾损伤是指由一些中药所致、具有不同临床表现和病理特征的一组疾病。
(一)药物性肾损伤的发病机制
药物性肾损伤根据给药种类、途径、剂量和疗程不同,发病机制各异,包括可直接引起肾小管损伤,或影响肾脏血流动力学变化导致肾缺血,或通过免疫机制引起过敏反应,或激活多种途径导致肾间质纤维化。中药相关肾损伤通常以肾小管及肾间质为主,亦可为肾小球及肾血管,少部分中药可引起全身其他脏器损伤而累及肾脏。
(二)药物性肾损伤的诊断依据
中药导致肾损伤的临床表现常缺乏特异性。目前尚无公认的药物相关肾损伤的诊断标准,也无特异性诊断标志物。中药相关肾损伤的诊断依据主要包含以下几方面:
1.用药史:发生肾脏损伤前有明确的药物使用史。需记录药物种类、剂量、疗程、用药与肾损伤发生的时间、停药后肾损伤恢复情况等。
2.易损人群:高龄或儿童患者;慢性肾脏病患者;慢性系统性疾病如高血压、心脑血管疾病、糖尿病、慢性肝病等;全身血容量下降或肾脏局部血流量下降者;过敏体质者;感染及危急重症患者;因其他疾病同时联用多种药物者。
3.临床表现:主要表现为血尿、蛋白尿、多尿或少尿、夜尿增多、腰痛、水肿、高血压等。也可见发热、皮疹、皮肤瘙痒、关节疼痛、乏力、食欲不振、恶心呕吐、贫血、心慌、气短等肾外表现。
4.实验室检查:尿常规及尿沉渣镜检;微量白蛋白尿;24小时尿蛋白定量;肾小球功能,如Scr、肾小球滤过率(GFR)、内生肌酐清除率(CCr)、Cystatin C等;肾小管功能,如尿NAG酶、尿β2-MG、尿α1-MG、RBP、尿渗透压、尿电解质等;肾脏B超等。
5.肾活检病理检查:表现为急性肾小管坏死、急性间质性肾炎、慢性肾小管间质病变、肾小球及肾血管病变。
6.停药后肾损伤变化情况:如停药后肾损伤恢复则更加支持药物相关肾损伤的诊断。
(三)药物性肾损伤的诊断与分级标准
药物性肾损伤根据损害部位、发病机制及临床表现,一般分为急性或慢性药物性肾损伤。急性药物性肾损伤包括急性肾损伤(acute kidney injury, AKI)和急性肾脏病(acute kidney disease, AKD);慢性药物性肾损伤多属慢性肾脏病(chronic kidney disease, CKD)的范畴。其诊断及严重程度分级标准可参照2012年改善全球肾脏病预后组织(kidney diseases improvement global outcome, KDIGO)制定的相关标准。
1.急性肾损伤
(1)AKI的诊断标准:符合以下情况之一:①48小时内Scr至少上升26.5µmol/L(0.3mg/dl);②Scr上升超过基础值的1.5倍,且确认或推测7天内发生;③尿量<0.5ml/kg/h,且持续6小时以上。单用尿量改变作为判断标准时,需要除外尿路梗阻及其他导致尿量减少的原因。
(2)AKI的分期标准(表1)
表1 2012年KIDGO关于AKI分期标准
分期
血清肌酐
尿量
1
Scr上升至基础值的1.5~1.9倍
或Scr上升≥0.3mg/dl(26.5µmol/L)
<0.5ml/kg/h,6~12小时
2
Scr上升至基础值的2.0~2.9倍
或GFR下降>50%
<0.5ml/kg/h,≥12小时
3
Scr上升至基础值的3倍以上
或Scr上升≥4mg/dl(353.6µmol/L)
或开始肾脏替代治疗
或年龄小于18岁的患者,GFR下降至<35ml/min/1.73m2
<0.3ml/kg/h, >24小时
或无尿≥12小时
(3)AKD的诊断标准:符合以下任何一项:①符合AKI的诊断标准;②3个月内在原来基础上,GFR下降35%或Scr上升50%;③GFR<60ml/min/1.73m2、<3个月。
2.慢性肾损伤
(1)CKD的诊断标准:①有肾损害标志,持续超过3个月。肾损害包括:蛋白尿>30mg/d;尿沉渣检查异常(如血尿、红细胞管型等);肾小管功能障碍导致的电解质等异常;肾脏病理检查异常;影像学检查发现肾脏结构异常;有肾移植史。②GFR<60ml/min/1.73m2、持续超过3个月。③GFR<90ml/min/1.73m2时,就是慢性肾衰竭的开始。
(2)CKD的分期标准(表2)
表2 2012年KIDGO关于CKD的分期标准
分期
描述
GFR(ml/min)
G1
正常或增高
≥90
G2
轻度肾功能下降
60~89
G3a
轻中度肾功能下降
45~59
G3b
中重度肾功能下降
30~44
G4
重度肾功能下降
15~29
G5
肾衰竭
<15
G5D
已经接受透析治疗
<15
(四)肾脏安全性评价指标
1.常规肾脏安全性评价指标
(1)尿常规及尿沉渣镜检。
(2)微量白蛋白尿(推荐使用即刻尿白蛋白与尿肌酐的比值,UACR)。
(3)肾小球功能:Scr和/或eGFR(推荐使用简化MDRD公式或CKD-EPI公式)。
(4)肾小管功能:尿NAG酶。
2.视情况增加的肾脏安全性评价指标
(1)药理毒理研究提示有肾毒性或处方中含有肾毒性报道的药物,早期临床试验观察到药物可能对肾脏有潜在毒性的,可增加血清Cystatin C、eGFR。
(2)临床试验期间中尿常规检查尿蛋白阳性、且1~2周后复查仍为阳性者,可增加24小时尿蛋白定量。
(3)临床试验期间尿沉渣镜检发现血尿、且48小时后复查仍有血尿者,可增加尿红细胞位相。
(4)临床试验期间连续2次尿NAG酶升高2倍及以上者,可增加其他肾小管功能检查指标如尿α1-MG、尿β2-MG、尿RBP等。
3.检测时点
(1)疗程≤1周者,治疗前、后各检测1次。
(2)疗程>1周者,治疗前、治疗后第1~2周各检测1次,以后每4周检测1次,12周后至少每12周检测1次,直至疗程结束。
(3)非临床安全性研究提示有肾毒性或处方中含有肾毒性报道的药物以及易损人群等,应当酌情增加检测频度。
(五)药物性肾损伤的处理原则
1.停止用药的指征
(1)急性肾损伤:参照2012年KDIGO关于AKI的诊断标准。
(2)3个月内Scr升高>50%,和/或GFR下降>35%。
2.在临床试验过程中如以下检测指标发现异常,应及时复查予以确认,必要时咨询肾病专家作进一步评估。
(1)尿NAG酶升高2倍及以上。
(2)其他肾小管功能检测指标如尿α1-MG、尿β2-MG、尿RBP等升高2倍及以上。
(3)新发生的尿蛋白、且24小时尿蛋白定量≥0.5g。
(4)新发生的尿微量白蛋白。
(5)新发生的血尿。
(6)血肌酐升高25%~50%。
十三、参考文献
1.《中药新药临床研究指导原则》(试行),中国医药科技出版社,2002年.
2.E1-E10 ICH:International Conference on Harmaonisation of Technical Requirements for Registration of Pharmaceuticals for Human use ICH Harmonised Tripartite Guaidline.
3.Danan G, Benichou C. Causality assessment of adverse reactions to drugs-I.A novel method based on the conclusions of international consensus meetings: application to drug-induced liver injuries. J Clin Epidemiol 1993; 46: 1323‐1330.
4.Tajiri K,Shimizu Y. Practical guidelines for diagnosis and early management of drug-induced liver injury[J].World J Gastroenterol,2008,14(44):6774-6785.
5.De Valle MB,Av Klinteberg V,Alem N,et al.Drug-induced liver injury in a Swedish University hospital outpatient hepatologyclinic[J].Aliment Pharmacol Ther,2006,24,1187-1195.
6.Larrey D. Epidemiology and individual susceptibility to adverse drug reactions affecting the liver. Seminars In Liver Dis. 2002;22(2):145-155.
7.International Consensus Meeting. Criteria of drug-induced liver disorders. J Hepatol 1990; 11:272-276.
8.Chalasani N P, Hayashi P H, Bonkovsky H L, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol, 2014, 109(7): 950-966.
9.Hayashi PH, Fontana RJ. Clinical features, diagnosis, and natural history of drug-induced liver injury. Semin Liver Dis, 2014, 34(2):134-144.
10.Leise MD, Poterucha JJ, Talwalkar JA. Drug-induced liver injury. Mayo Clin Proc, 2014, 89(1):95-106.
11.Fontana RJ, Seeff LB, Andrade RJ,et al. Standardization of nomenclature and causality assessment in drug-induced liver injury: summary of a clinical research workshop. Hepotology,2010,52(2):730-742.
12.Zimmerman, HJ, 1978,Drug-Induced Liver Disease, in:Hepatotoxicity, The Adverse Effects of Drugs and Other Chemicas on the Liver,1st ed.pp.351-3, Appleton-Centu ry -Crofts, New York.
13.Zimmerman, HJ, 1999, Drug-Induced Liver Disease, in:Hepatotoxicity, The Adverse Effects of Drugs and Other Chemicals on the Liver, 2nd ed.pp.428-33, Lippincott Williams & Wilkins, Philadelphia.
14.http://www.fda.gov/cher/guidelines.htm
15.Temple,R,2001,Hepatotoxicity Through the Years: Impact on the FDA, presented/12/2001, http://www.fda.gov/downloads/
Drugs/ScienceResearch/ResearchAreas /ucm122149.pdf.
16.Reuben, A, 2004, Hy’s Law, Hepatology, 39(2):574-8.
17.Guidance for Industry Drug-Induced Liver Injury:
Premarketing Clinical Evaluation. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER),Center for Biologics Evaluation and Research (CBER). July 2009, Drug Safety (http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm. )
18.The Criteria Committee of the New York Heart Association. Nomenclature and Criteria for Diagnosis of Diseases of the Heart and Great Vessels. 9th ed. Boston, Mass: Little, Brown & Co; 1994:253-256.
19.ftp://ftp1.nci.nih.gov/pub/cacore/EVS/CTCAE/Archive/CTCAE_4.0_2009-05-29_QuickReference_8.5x11.pdf.
20.http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf
21.http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073161.pdf
22.陈冀莹. 心肌损伤标记物的应用及临床意义. 国外医学放射医学核医学分册 2004; 28: 216-219.
23.Kilickap S, Barista I, Akgul E, et al. Ctnt can be a useful marker for early detection of anthracycline cardiotoxicity. Annals of Oncol 2002; 13: 699.
24.Maisel S, Koon J, Krishnaswamy P, et al. Utility of BNP as a rapid, point of care test for screening patients undergoing echocardiography to determine left ventricular dysfunction. Am Heart J 2001; 14(30): 367。
25.Lee HS, Son CB, Shin SH, et al. Clinical correlation between brain natriuretic peptide and anthracyclin-induced cardiac toxicity. Cancer Res Treat 2008; 40(3): 121-126.
26.Okumura H, Iuchi K, Yoshida T, et al. Brain natriuretic peptide is a predictor of anthracyclin-induced cardiotoxicity. Acta Haematol 2000; 104(4): 158-163.
27.张思洁,崔彦芝. 脑钠肽检测在蒽环类药物化疗心脏毒性患者中的意义. 疑难病杂志 2011; 10(6): 444-445.
28.Feola M, Garrone O, Occelli M, et al. Cardiotoxicity after anthracycline chemotherapy in breast carcinoma: Effects on left ventricular ejection fraction, troponin I and brain natriuretic peptide. Int J Cardiol 2011; 148: 194-198.
29.Marchandise B, Schroeder E, Bosly A, et al. Early detection of doxorubicin cardiotoxicity: interest of Doppler echocardiographic analysis of left ventricular filling dynamics. Am Heart J. 1989; 118: 92-98.
十四、附录
急性药物性肝损伤因果关系评价(RUCAM)
肝 细 胞 型
胆汁淤积或混合型
评 价
1.服药至发病时间
不相关
反应发生在开始服药前或停药后超过
15天*
反应发生在开始服药前或停药后超过
30天*
无相关性
未知
无法获得服药至发病时间 无法获得服药至发病时间
无法评价
初次治疗
随后的治疗
初次治疗
随后的治疗
计 分
从服药开始
提示
可疑
从停药开始
可疑
5~90天
<5天或>90天
≤15天
1~15天
>15天
≤15天
5~90天
<5天或>90天
≤30天
1~90天
>90天
≤30天
+2
+1
+1
2.病程
ALT峰值与正常上限之间的差值
ALP或TBil峰值与正常上限之间的差值
停药后
高度提示
提示
可疑
无结论
与药物作用相反
如果药物仍在使用
无结论
8天内降低>50%
30天内降低≥50%
在30天后不适用
没有相关资料或在30天后下降≥50%
30天后下降<50%或再升高
所有情况
不适用
180天内下降≥50%
180天内下降<50%
不变、上升或没有资料
不适用
所有情况
+3
+2
+1
0
-2
0
3.危险因子
酒 精
酒精或怀孕
有
无
+1
0
+1
0
年龄≥55岁
年龄<55岁
4.伴随用药
无或伴随用药使用时间与发病时间不符合
伴随用药使用时间与发病时间相符合
已知伴随用药有肝毒性且使用时间与发病时间相符合
有证据表明伴随用药致肝损伤(再用药反应或有价值的检测)
0
-1
-2
-3
5.除外其他原因
(1)近期感染过甲肝病毒(anti-HAV-IgM) 或乙肝病毒( anti-HBc-IgM) 或丙肝病毒(anti-HCV) 或有其他非甲非乙型肝炎感染的证据;胆道梗阻(B超);酗酒(AST/ALT≥2)。近期(2周内)有低血压、休克或肝缺血史。
(2)有重要疾病并发症;临床和/或实验室提示CMV、EBV或疱疹病毒感染
●所有原因,包括(1)和(2)完全排除
●(1)中所有原因被排除
●(1)中4~5个原因被排除
●(1)中少于4个原因被排除
●高度怀疑非药物因素
+2
+1
0
-2
-3
6.药物既往肝损伤的报告
产品说明中有肝毒性报告
有文献报道但产品说明中无相关信息
尚无肝毒性报道
+2
+1
0
7.再用药反应
阳性
可疑
阴性
未做或不可判断
单用该药物ALT升高≥2×ULN
与首次发生肝损伤时的合并用药一起给药致ALT升高≥2×ULN
再用同样药物ALT仍在正常范围
其他状况
单用该药物ALP或TBil升高≥2×ULN
与首次发生肝损伤时的合并用药一起给药致ALP 或TBil升高≥2×ULN
再用同样药物ALP或TBil仍在正常范围
其他状况
+3
+1
-2
0
注:* 慢代谢型药物除外。最后判断:>8分,非常可能;6~8分,很可能;3~5分,可能;1~2分,不象;≤0分,无关。
(一)试验目的
(二)临床试验设计方法
(三)受试者的选择与退出
(四)对照的设置
(五)样本量
(六)给药方案
(七)基线和均衡性
(八)有效性指标观测与评价
(九)安全性指标观测与评价
(十)随访



20151103中药新药临床研究一般原则
2022年3月18日发布

商务拓展部(药品&器械)袁经理
电话:010-83659936 18618362640
邮箱:ybkj045@126.com
人力资源部主任
电话:13146546661
邮箱:ybkj010@126.com
商务总监(药品&器械)薛总
电话:13810708945
邮箱:ybkj027@126.com








◆丰富的临床试验服务经验:
◆熟悉政策法规及审评动态:
◆良好的专家关系:
◆大量临床医院的资源:
◆专业的数据管理:
CRO:Contract Research Organization, 出现于上世纪80年代,一种学术性或商业性的科学机构。申办者可委托其执行临床试验中的某些工作和任务,此种委托必须作出书面规定,其目的是通过合同形式向制药企业提供新药临床服务的专业公司。CRO 可以作为制药企业的一种可借用的外部资源,可在短时间内迅速组织起一个具有高度专业化的和具有丰富临床经验的临床队伍,并能降低整个制药企业的管理费用。
 了解更多
了解更多
了解更多
了解更多








我公司承接国内外厂家或公司的委托,从事药物、医疗器械临床代理服务,以下是我公司服务过的部分项目的名称。









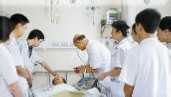





北京远博医药CRO成立于2005年,为医疗企业提供临床试验、项目申报、项目管理和信息咨询等的专业化外包服务(CRO服务),主要为专业化服务内容包括:新药(化药、中药和生物制品)的Ⅰ~Ⅳ临床试验、药代动力学(PK)、生物等效性(BE)、医疗器械的临床试验、上市后再评价、中药保护临床试验、药物经济学等项目的监查及项目管理。
专业化人才优势
绝大多数员工拥有专科以上学历
高标准质量管理体系
必须通过国家药监局高级研修学院的GCP培训并获得证书
完善的培训系统
注重对员工的技术培训,积极参加国家药监总局、药审中心、器械审评中心及各学术团体举办的专业培训及会议;
◆丰富的临床试验服务经验:
◆熟悉政策法规及审评动态:
◆良好的专家关系:
◆大量临床医院的资源:
◆专业的数据管理:
◆优秀而稳定的团队:
◆完善的标准操作流程:
◆独立的质量保障部门:
◆灵活多样的服务方式:






































































































 预约上门洽谈
免费项目咨询
预约上门洽谈
免费项目咨询CRO

北京远博医药

临床试验、一致性评价、BE研究和技术服务的专家
专业为医药企业提供一体化的专业技术服务


临床试验优先选择的CRO
18618362640
010-83659936
全国客服热线